
【论文标题】A deep learning-based framework for estimating fine-scale germline mutation rates
【作者团队】, ,
【发表时间】2021/10/27
【机 构】中山大学
【论文链接】https://doi.org/10.1101/2021.10.25.465689
【代码链接】https://github.com/CaiLiLab/MuRaL
胚胎突变率对遗传和进化分析至关重要。然而,由于观察到的突变相对较少,而且预测因子和突变率之间的关系错综复杂,估计整个基因组的精细尺度突变率是一个巨大的挑战。在这里,本文提出了MuRaL,即突变率学习器,一个基于深度学习预训练的框架,只使用基因组序列作为输入来预测细尺度的突变率。利用人类种系突变体进行综合评估,本文表明MuRaL比目前最先进的方法实现了更好的预测性能。此外,MuRaL可以用相对较少的训练突变和中等数量的测序个体建立模型。它可以利用迁移学习,以更少的训练数据和时间建立模型。本文应用MuRaL为四个物种--智人、猕猴、拟南芥和黑腹果蝇产生全基因组的突变率谱,证明了它的高度适用性。生成的突变率图谱和开源软件可以大大促进相关研究。
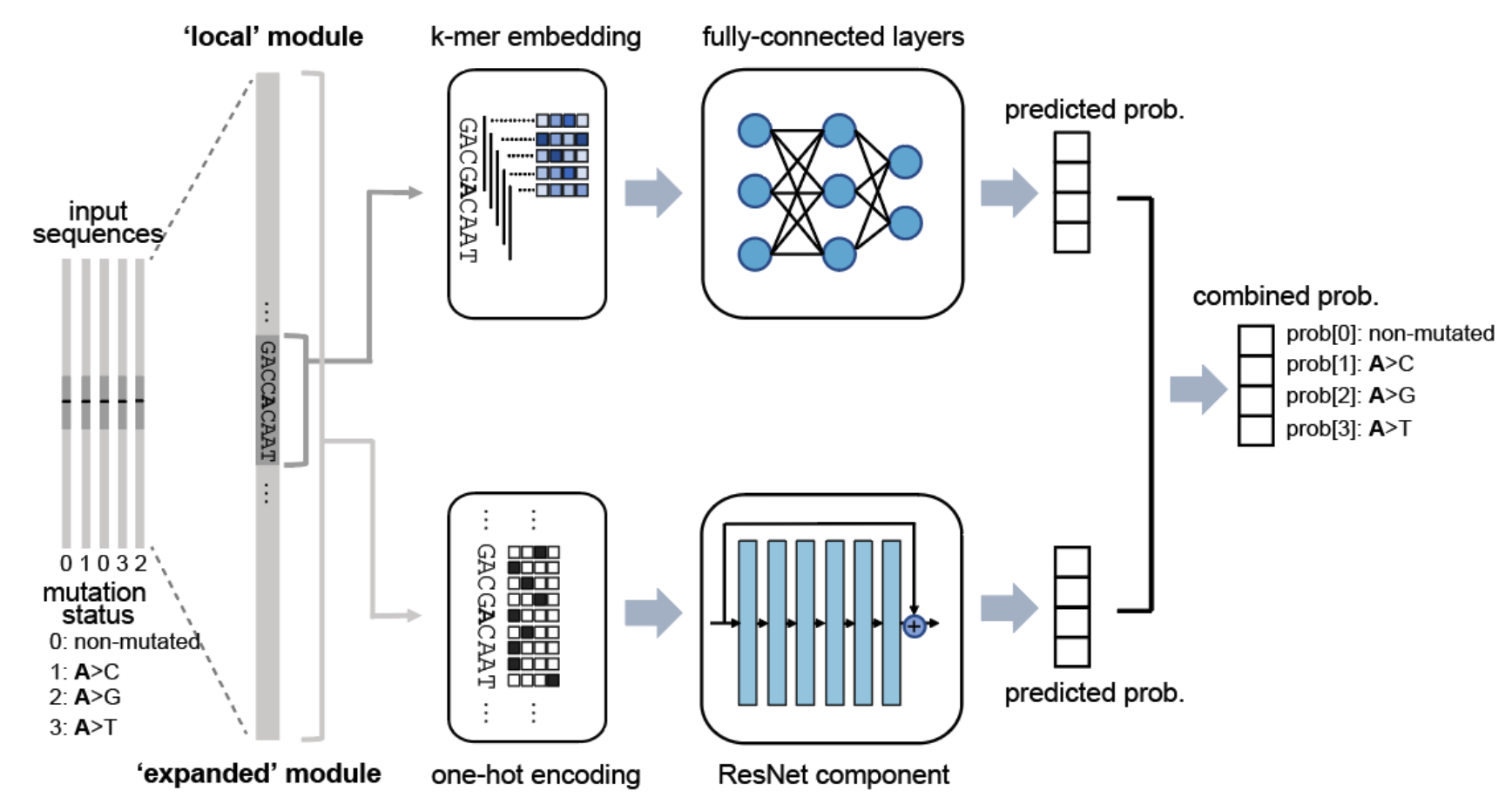
上图展示了MuRaL模型框架,该模型由一个 "局部 "模块和一个 "扩展 "模块组成。在 "局部 "模块中,焦点核苷酸的输入序列(如图中的粗体 "A")被分割成重叠的K-mers,然后由嵌入层映射成多维向量。多维向量被串联起来,并传递给三个全连接(FC)层。“局部”模块的输出是由softmax函数在四个预测类别上产生的概率分布--非突变或三种可能的替代突变之一(A>C、A>G和A>T)。在 "扩展 "模块中,扩展区域的输入序列进行one-hot编码。one-hot编码的矩阵被视为具有四个通道的一维数据,并传递给ResNet组件。一个额外的FC层和ResNet组件之后的softmax函数在四个预测的类别上产生一个概率分布,就像在 "局部 "模块中一样。“局部 "和 "扩展 "模块的概率使用相等的权重(即0.5*Plocal + 0.5*Pexpanded)进行组合,产生组合概率。
拥有这两个模块的主要原因是,局部和远端序列可能以不同的方式对焦点核苷酸的突变性做出贡献,因此它们中的信号可能会被不同的网络架构更好地学习。
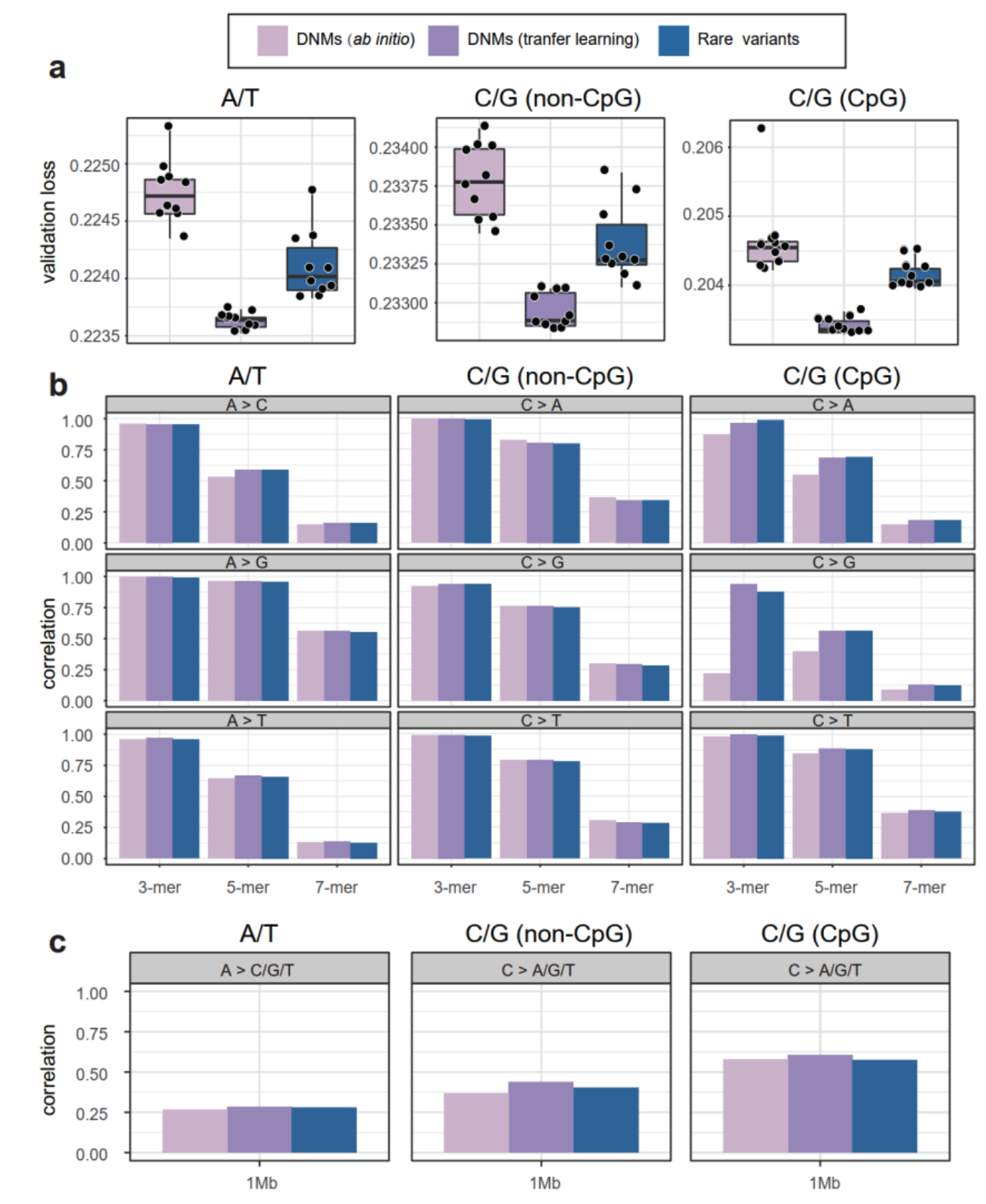
上图展示了训练de novo 突变(DNM)和迁移学习的结果
为了研究MuRaL框架中迁移学习的有效性,本文用相同的DNM来训练迁移学习模型,使用上述稀有变量模型的预训练权重进行模型初始化。通过独立的验证DNM,本文发现具有迁移学习的模型比没有迁移学习的模型(非初始DNM模型;a图)取得了明显的验证损失。迁移学习模型还显示出更好的k-mer和区域突变率的相关性,这是用DNMs作为观察到的突变计算出来的(b,c图)。
(a) 三类模型在验证DNM上的平均验证损失。DNM ab initio模型、DNM迁移学习模型和稀有变异模型。对于每个模型,十次试验中每次的最低平均交叉熵损失的boxplot。
(b) 根据人类Chr1上的单核苷酸突变率预测,不同突变类型的3、5和7-mer突变率的相关性。对于a图中的每个模型,具有最低验证损失的最佳试验被用于预测Chr1上的突变率。
(c) Chr1上1Mb大小的区域突变率相关性。多个突变类型(如A>C/A>G/A>T)的预测突变率被聚集起来计算区域相关性,因为一些突变类型在数据中观察到的DNM非常少,所以没有评估较小的bin大小。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢