什么是水平基因转移?我们熟悉的从父母遗传给孩子一般被称为垂直基因转移;水平基因转移,则是指的是一个或多个基因,通过父母遗传以外的方式进行传递。
水平基因转移常见于细菌之中,有些细菌主动同周围的细菌发生接合作用并共享基因,从而实现基因转移。即使它们是完全不同的种类,一些细菌收集从其他细胞释放出来的基因并将它们收为己用。如果外源基因适应良好,在增殖时细菌会将外源基因传递给后代;假如这个外源基因是抗生素抗性基因,将会给疾控带来了非常大的困扰。
系统发育距离、共享生态学和基因组约束通常被认为是控制水平基因转移(HGT)的关键驱动因素,尽管它们的相对贡献尚不清楚。
康奈尔大学的研究人员将机器学习算法应用于一组精选的不同细菌基因组,以梳理特定功能特征对 HGT 事件的重要性。
研究人员发现功能性内容准确预测HGT网络,涉及抗生素抗性基因(ARG)的转移的性能进一步提高,突出了HGT机制、生态位特异性和代谢功能的重要性。发现尚未检测到的高概率 ARG 转移事件,几乎是人类相关细菌所独有的。该方法在预测病原体的 HGT 网络方面是可靠的,包括鲍曼不动杆菌和大肠杆菌,以及在局部环境中,例如个人的肠道微生物组。
该研究以「Functions predict horizontal gene transfer and the emergence of antibiotic resistance」为题,于 2021 年 10 月 22 日发布在《Science Advances》。
水平基因转移(Horizontal Gene Transfer,HGT)是一种普遍的进化过程,导致基因在不同的原核谱系之间分布。尽管这一过程塑造了微生物的古老进化,但最近的转移事件是抗生素或金属抗性基因、毒力因子和其他对人类感染过程具有深远影响的特征传播的基础。完整原核基因组的广泛测序使得进行系统的、基因组规模的比较以识别基因组内的 HGT 区域和描绘与 HGT 率相关的特征成为可能,尽管这些努力未能预测基因在整个基因组中的扩散。
先前研究基因流动性的努力报告说,与远亲生物相比,系统发育相关生物中以及具有共享 GC 含量和 kmer 含量或甲基化模式的生物之间的 HGT 发生率更高。基因组内容,如特定质粒复制子和动员机制或共享噬菌体结构蛋白的存在,也决定了微生物物种间的基因流动,而限制性修饰基因、CRISPR-Cas9适应性免疫系统和毒素抗毒素系统的存在则是基因流动的障碍。细菌基因组或移动元件固有的遗传因素有利于密切相关的生物体之间的转移,因为它们与天然分子机制或更大的序列同源性具有更大的兼容性。
跨不同空间尺度的生态环境也影响 HGT 率,丰富了同一环境(例如,海洋、宿主相关和土壤)内发现的生物体或从多个宿主的同一身体部位分离的生物体之间的 HGT。许多生态特征是垂直遗传的,因此映射到物理化学梯度或环境资源斑块上。然而,邻居之间移动功能特征的获取和维护增加了生态特定适应和微生物物种形成的潜力。这种环境选择可以在不同饮食人群的肠道微生物组中移动遗传元件的含量,以及受到不同抗生素负担的牲畜微生物组中移动抗生素抗性基因 (ARG) 的组成中观察到。这表明除了兼容性之外,HGT 还需要接近度。
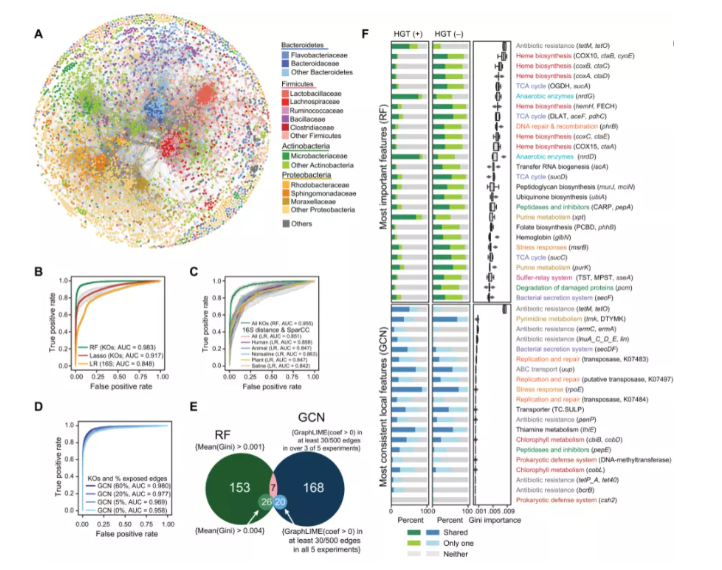
尽管认识到各种因素对整体 HGT 率的影响,但很难对包含和权衡这些宏观和微观选择压力的 HGT 进行全面了解。假设功能基因含量将是 HGT 的重要决定因素,因为基因含量同时反映系统发育、基因组和生态因素。为了测试这一点,研究人员利用公开的基因组数据库创建了一个HGT事件网络。该网络包括基因组特异性因素,如功能内容(节点特征)和相关因素,如系统发育距离和共现性(边缘特征)。我们实施了几种机器学习方法,即逻辑回归(LR)、随机森林(RF)和图卷积神经网络(GCN)模型,以量化它们对HGT的影响,因为它们的多功能性、它们对基因组学和细菌表型的实用性,以及他们预测网络中多维链接的能力。此外,这些方法允许使用节点和边缘效应来解释网络中事件和特征的非独立性,这在解析 HGT 事件的复杂病因时很重要。
该团队构建了一个观察到的基因转移事件的无向网络。在从几个公共数据库下载基因组后,特别注意去除这些基因组中可能被错误注释为 HGT 的任何潜在污染物(宿主或载体传播的),以及由CheckM确定的完整性较低(<90%)或污染分数较高(>5%)的任何基因组。
这产生了一个由 12,518 个分离和测序的基因组组成的数据集,代表 10,500 多个细菌物种,从公开可用的数据集中获得。为了减少抽样偏差,研究人员为每个物种最多选择了三个分离株或 97% 16S 核糖体 RNA (rRNA) 相似性簇。由于在这种大小的基因组上应用基于系统发育的方法进行 HGT 检测的计算限制,以及在大规模识别密切相关生物之间最近转移的挑战;研究人员使用先前审查过的启发式方法来识别参与 HGT 的生物。
HGT 阳性边缘被定义为远亲生物(其 16S rRNA 的序列相似性低于 97%)之间的那些,它们具有至少 500 个碱基对 (bp) 的几乎相同(99% 或更高的序列相似性)DNA 区域。最终的 HGT 网络是稀疏的,由 6566 个基因组中观察到的 147,889 个 HGT 事件或大约 7830 万个成对比较的 0.189% 组成。
利基特异性、代谢和移动性状对于预测 HGT 很重要
鉴于 RF 和 GCN 模型的性能,研究人员试图检查利用哪些功能特征来生成 HGT 的预测。虽然可以从 RF 模型中轻松提取特征重要性,但 GCN 的两个隐藏层中使用的特征不太容易立即解释。该团队采用了 GraphLIME 方法,该方法通过检查网络中特定节点的特征来衡量特征对网络中特定节点的重要性,以评估边缘预测。
在整个实验中始终观察到一个特征子集。尽管 RF 和 GCN 模型的结构和特征选择方法存在很大差异,但研究人员发现用于预测 HGT 的模型各自重要且一致的 KO 之间存在重叠。GCN 中的重要特征可能涉及在紧密连接的局部组件中促进/抑制 HGT 的功能,而 RF 选择在系统发育中具有广泛重要性的特征。
门间(Interphylum) HGT 事件特别重要,因为它们可能导致在 HGT 与共生生物发生后,病原生物最近出现抗生素耐药性。远亲生物之间的「长距离」HGT 事件被认为是罕见的,除非在某些极端环境中的物种之间,例如在富含有机物的白蚁或反刍动物肠道中的嗜盐生物、嗜热生物、糖解生物或发酵生物之间。
然而,实验支持在放线菌和变形菌之间转移 ARG 的可行性。数据集中最近的长距离 HGT 事件仅代表 11.87%(147,889 个中的 17,561 个)HGT 阳性边缘,并且单独使用 16S rRNA 距离进行预测是有问题的(平均 AUROC = 0.499)。
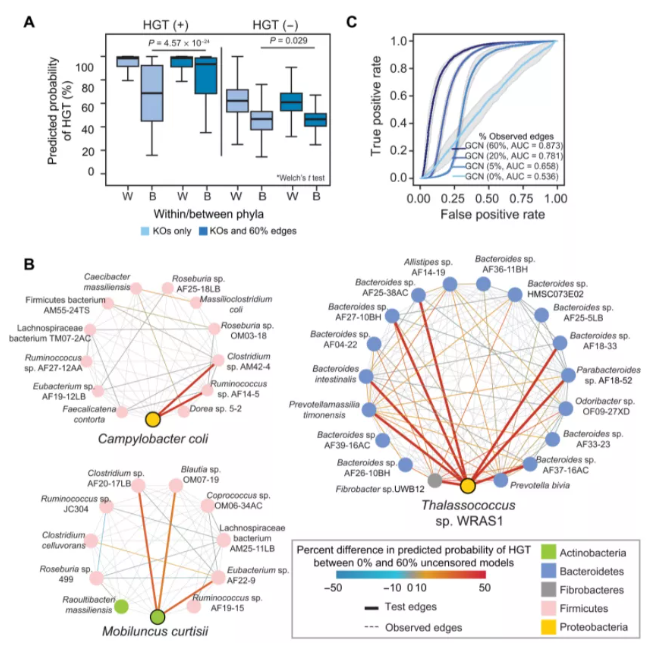
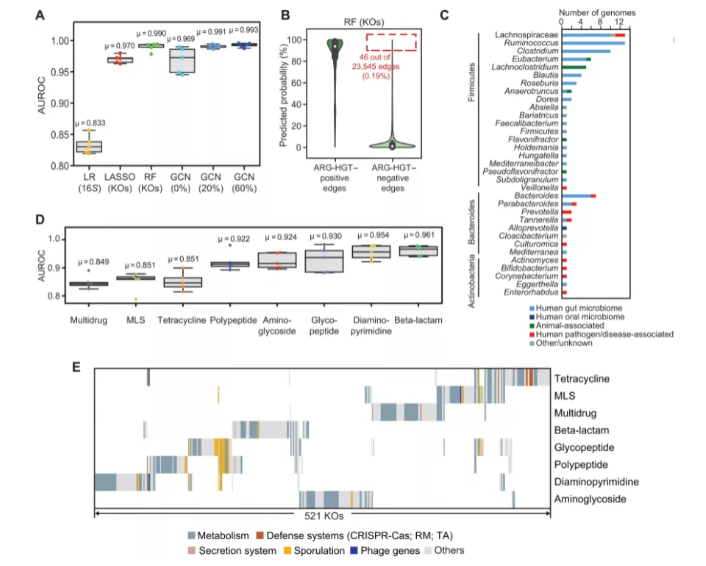
鉴于新出现的抗生素耐药性的临床重要性,研究人员评估了预测时特别涉及一种或多种 ARG 转移的能力;这些转移占观察到的 HGT 事件的 43.63% 和所有门间转移的 47.44%。研究人员排除了 645 个 KO 的输入 KO 矩阵,这些 KO 矩阵与已知的 ARG 甚至具有模糊的相似性。
尽管边缘较少,但 HGT 预测提高到近乎完美的准确度(RF:平均 AUROC = 0.990;具有 60% 未删失边缘的 GCN,平均 AUROC = 0.993)并且重要特征在所有转移训练的模型中保持基本一致。
鉴于这种性能,研究人员检查了测试数据中的稀有边缘(23,545 个边缘中的 46 个,或 0.2%),这些边缘实现了 ARG-HGT 的高预测概率(超过 0.9),但没有检测到转移。这些边缘几乎是厚壁菌门、拟杆菌门和放线菌门的人类相关肠道和口腔微生物组成员所独有的;涉及几种病原体;并且不同于随机排列的 HGT 负边缘样本。
这些结果不仅突出了人类微生物组的混杂性质,而且突出了 ARG 进一步传播到其他分类群的尚未发现的潜力。
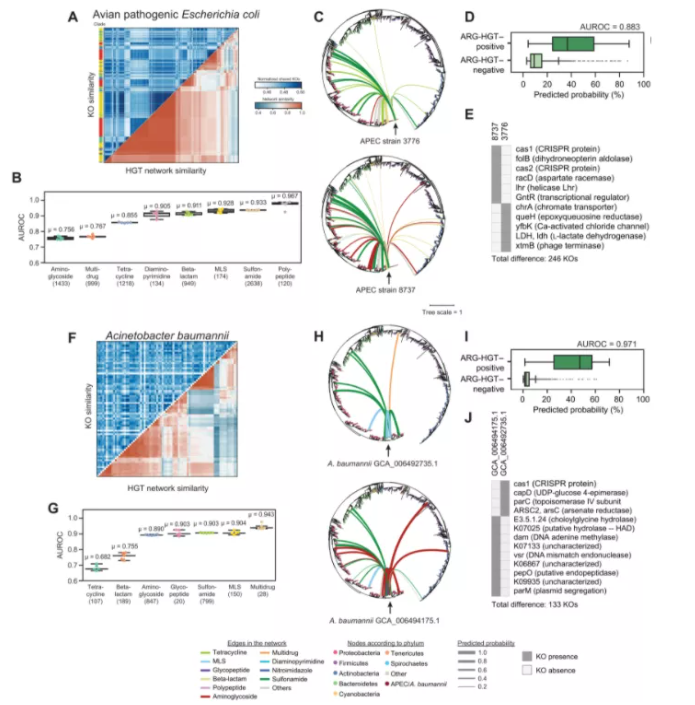
这项工作的前景在于它能够预测病原体之间的潜在传播。作为概念证明,研究人员使用原始数据集回顾性分析了病原分离物集合的 HGT 网络。
首先,分析了近 40 年来收集的 433 种不同禽类致病性大肠杆菌菌株的集合。同一系统发育进化枝内的分离株在观察到的 HGT 网络方面有所不同。
研究人员在所有 ARG 类别中都实现了可靠的预测(平均 AUROC 范围从 0.756 到 0.967)。说明了两个菌株的预测网络,以及区分这两个混杂基因组的重要特征的存在和缺失,揭示了功能含量相对较少差异对其的影响 各自的网络。这反映了代谢基因的单一突变导致转录组和抗生素敏感性发生巨大变化的观察结果。
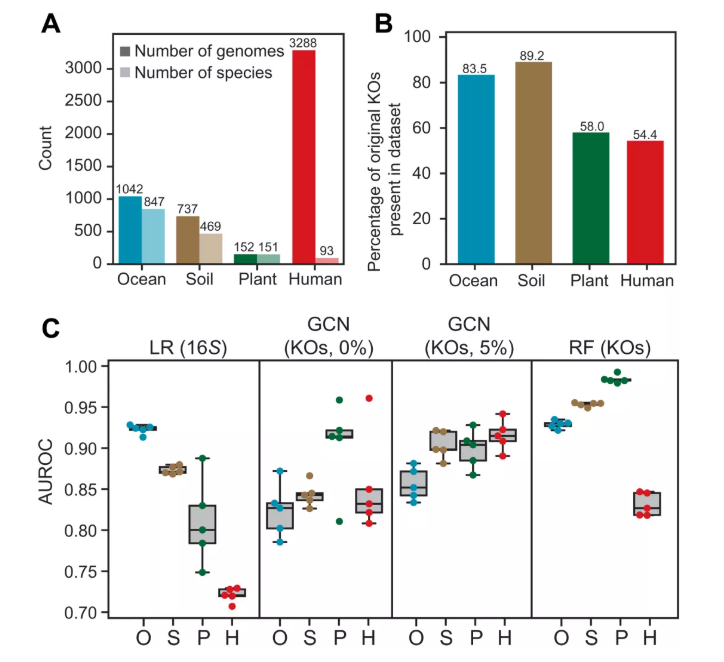
鉴于选择可能在不同的空间尺度上起作用,包括在单个宿主的水平上,研究人员试图确定该方法是否对来自单个环境或由单个实验室或联盟生产的相对较小的数据集内的 HGT 预测有效 ,其中移动遗传元件的分散,而不是选择,可能会支配信号。研究人员确定了来自各种环境 [海洋、土壤、植物根和人体肠道] 的四个正交分离物数据集,并应用于在 12,000 多个分离物上构建的原始模型,来预测 HGT。海洋数据集包含 847 个物种,而人类肠道数据集包含 3288 个高质量基因组,尽管仅代表 93 个物种。
不同数据集的 HGT 发生率差异很大:人类肠道内的 HGT 率最高 (17.92%),正如之前观察到的,海洋和土壤数据集低 60 倍以上(分别为 0.279 和 0.237 %),并且在植物根数据集中仅观察到 26 个 HGT 事件。由于仅表示原始数据集中 KO 的一个子集,在人类肠道数据集中只有 54.4%,研究人员甚至质疑他们的模型是否足以捕获这些数据集中的 HGT 网络。
HGT 的预测对于小型生态特定数据集是准确的。
HGT 的模式可以从大规模 HGT 网络中提取。使用机器学习,确定了定义最近 HGT 事件的特定原核功能,深入了解 HGT 各种驱动因素的相对贡献,包括利基特定属性、HGT 的机械障碍以及与转移基因子集相关的特征。研究人员的分析揭示了一系列可能的补偿和适应性功能;这些功能可能使生物体能够适应与基因获取相关的细胞应激,可能在移动遗传元件整合的热点遗传连锁,或涉及每个抗生素类别的ARG的功能或在移动元件上编码的其他功能。
机器学习算法对训练数据中的偏差很敏感。尽管选择具有代表性的数据集,但该方法对输入数据的质量很敏感(即,宏基因组组装的基因组过于嘈杂,因此被排除在外),并且必然存在盲点。增强培养组学的进一步改进,以及宏基因组和单细胞组装的持续改进,将使微生物组范围的 HGT 网络预测成为可能。同样,适用于最近时间尺度或可大规模应用的密切相关菌株之间的系统发育重建,可能会提供更高的分辨率和 HGT 事件的方向性。
令人惊讶的是,最近 HGT 事件的可预测性不仅在最广泛的范围内、跨门和生物群落是显而易见的,而且还扩展到特定环境,例如个体的肠道或植物的根茎,甚至在单个致病物种进化枝内, 表明选择的特征优于随机抽样。
最近 HGT 事件的可预测性让研究人员更好地了解细菌对快速变化的条件的适应,例如由抗生素的人为传播带来的条件。这开启了量化病原体和微生物组成分之间 HGT 风险的可能性,这些风险导致新型抗生素抗性菌株的出现和 ARG 库在局部环境中的扩张。该框架可用于改进用于改造微生物组的移动遗传元件的设计,或告知通过治愈或消除质粒或抑制结合来减少 ARG 负担的策略。内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢