分子的结构动力学由潜在的势能格局决定。锥形交叉点是连接原本分开的势能面的漏斗。近一个世纪前,圆锥形交叉点已经是科学界备受关注的主题。
在生物学中,它们在视觉、光合作用和 DNA 稳定性方面起着关键作用。用于检查锥形交叉点的准确理论方法目前仅限于小分子。实验研究受到所需时间分辨率和灵敏度的限制。
因此,当前对锥形交叉点的结构动力学理解仅限于具有大约 10 个原子的简单分子,时间尺度约为 100 fs 或更长。光谱可以实现更好的时间分辨率,但提供间接结构信息。
在这里,威斯康星大学与汉堡大学的研究人员合作,展示了光敏黄色蛋白质(一种 2,000 原子蛋白质)穿过锥形交叉点的几飞秒(fs,千万亿分之一秒)、原子分辨率的视频。
这些通过机器学习从实验数据中提取的视频,揭示了通过锥形交叉点去激发的动态轨迹,产生了控制去激发过程的锥形交叉点的关键参数,并阐明了所涉及的电子势能面的形貌。
该研究以「Few-fs resolution of a photoactive protein traversing a conical intersection」为题,于 2021 年 11 月 3 日发布在《Nature》。
作为分子几何学函数的分子电子的量子力学能量,产生了用于原子核运动的有效势能面。当有 d 个核自由度时,势能面(PES)是 d 维的。在所谓的波恩-奥本海默(BO)近似中,电子自由度和核自由度是分开处理的。当两个 PES 接触时,BO 近似不再有效。
锥形交叉点是这种势能简并的区域,形成一个(d – 2)维流形,在参与的电子态之间具有发散的、非 BO 耦合。由此产生的电子自由度和振动自由度的强烈混合开辟了一条途径,通过该途径,分子几何形状的动态变化可以导致从一种电子状态过渡到另一种电子状态。由于这会引起激发态的超快、非辐射弛豫,因此锥形交叉点在自然界中的许多过程中起着重要作用。光活性黄色蛋白(PYP)和视黄醛中对香豆酸发色团的反式到顺式异构化是主要例子。
处理耦合电子和振动动力学的准确理论方法目前仅限于小分子。这种模拟的质量——使用量子或经典的核运动处理——取决于所涉及的 PES 的精确表征和复杂性。例如,PYP 由 2,289 个原子组成,具有 6,861 个振动自由度。这种复杂程度使得严格的第一性原理电子结构计算,在可预见的未来变得不可行。
最先进的密度泛函理论可以应用于大小相当的分子,但还不能提供可靠的化学精度,特别是对于锥形交叉点。即使可以准确地求解单个分子几何的电子薛定谔方程,作为一个整体,对势能景观进行充分采样所需的分子几何总数,随着自由度的数量呈指数增长。
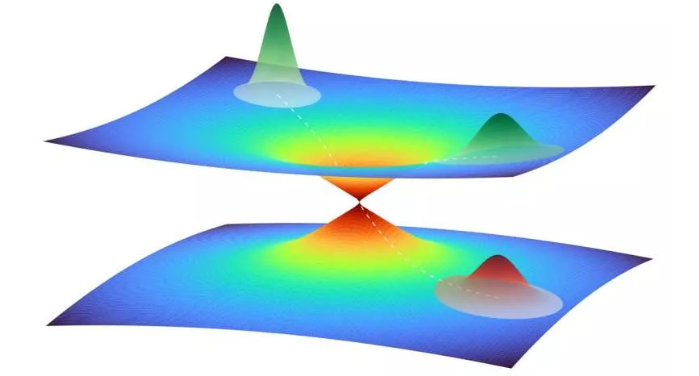
两个势能面之间锥形交叉点附近的量子波包的图示。
在实验上,通过锥形交叉点进行电子切换的结构-动力学模式的结论性观察,仍然难以捉摸。光泵-探针光谱提供了具有飞秒时间分辨率的电子态布居动力学信息,但它不提供对系统结构特性的直接访问。同样,时间分辨衍射技术,如超快电子衍射(UED)和时间分辨 X 射线衍射,到目前为止,缺乏通过锥形交叉点进行去激发所需的时间分辨率。近期,有研究团队将 UED 实验与广泛、复杂的从头计算模拟相结合,完成了时间分辨率为 150 fs 的 11 原子分子中锥形交叉诱导动力学的结构表征。
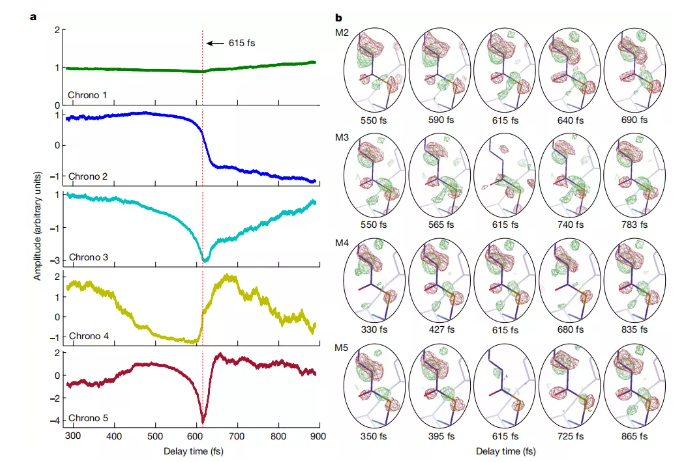
动态模式的演变作为泵 - 探针延迟时间的函数。
「为了准确了解自然界中的生化过程,例如某些细菌的光合作用,了解事件的详细顺序非常重要。」该团队的研究人员 Santra 说,「当光照射光敏蛋白质时,它们的空间结构会发生变化,这种结构变化决定了蛋白质在自然界中的作用。」
然而,直到现在,几乎不可能追踪结构变化发生的确切顺序。只有反应前后分子的初始和最终状态才能用理论术语来确定和解释。「但我们并不确切知道两者之间的能量和形状是如何变化的。」Santra 说,「这就像看到有人折叠了他们的手,但你看不到他们交叉手指这样做。」
一只手足够大,运动速度也足够慢,我们可以用眼睛跟随它,但在观察分子时,事情就没有那么容易了。分子的能量状态可以使用光谱学非常精确地确定;例如来自 X 射线激光器的明亮 X 射线可用于分析分子的形状。X 射线的极短波长意味着它们可以解析非常小的空间结构,例如分子内原子的位置。然而,结果不是像照片一样的图像,而是一个特征干涉图案,可以用来推断创建它的空间结构。
由于分子水平上的运动极其迅速,因此科学家们必须使用极短的 X 射线脉冲来防止图像模糊。只有随着 X 射线激光器的出现,才有可能产生足够明亮和短的 X 射线脉冲来捕捉这些动力学。然而,由于分子动力学发生在量子物理领域,物理定律偏离了我们的日常经验,因此只能借助量子物理分析来解释测量结果。
需要考虑光活性蛋白质的一个特殊特征:入射光激发它们的电子壳层进入更高的量子态,这会导致分子形状的初始变化。这种形状的变化反过来会导致激发态和基态量子态相互重叠。在由此产生的量子跳跃中,激发态恢复到基态,由此分子的形状最初保持不变。因此,量子态之间的锥形交叉为处于量子力学基态的蛋白质的新空间结构开辟了道路。
由 Santra 和 Ourmazd 领导的团队现在首次成功解开了这种锥形交叉点处光活性蛋白的结构动力学。他们通过机器学习来做到这一点,因为对动力学的完整描述实际上需要考虑所有涉及的粒子的每一种可能的运动。同样这也会产生无法解决的高维度的方程。
「我们研究的光敏黄色蛋白质由大约 2000 个原子组成。」DESY 首席科学家兼汉堡大学物理学教授 Santra 解释说,「由于每个原子基本上都可以在所有三个空间维度上自由移动,因此总共有 6000 种移动选项。这导致了一个具有 6000 个维度的量子力学方程——即使是当今最强大的计算机也无法解决。」
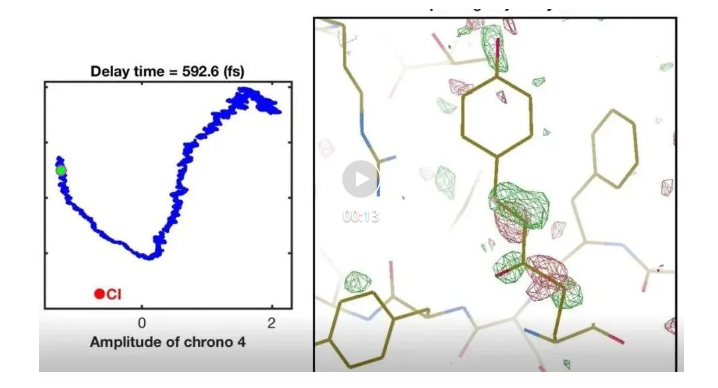
差异电子密度视频沿模式4/模式5的轨迹
然而,基于机器学习的计算机分析能够识别复杂分子中原子集体运动的模式。「这就像手在移动:在那里,我们也不是单独观察每个原子,而是观察它们的集体运动。」Santra 解释说。与集体运动的可能性显而易见的手不同,在分子的原子中识别这些选项并不容易。即便如此,使用这种技术,计算机能够将大约 6000 个维度减少到四个。通过展示这种新方法,Santra 的团队还首次能够表征由数千个原子组成的复杂分子中量子态的锥形交叉点。
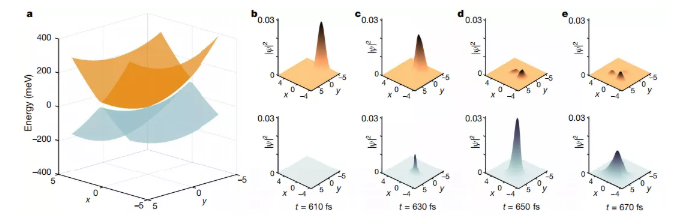
详细的计算显示了这个锥形交叉点是如何在四维空间中形成的,以及光敏黄色蛋白在被光激发后如何通过它回到初始状态。科学家们现在可以以几十飞秒为单位描述这个过程,从而加深对光敏过程的理解。
「因此,量子物理学为生物系统提供了新的见解,生物学为量子力学方法提供了新的思路。」Santra 说, 「这两个领域在这个过程中相互促进。」
第一,可以从现有的时间分辨晶体学数据中,以原子空间分辨率和几飞秒时间分辨率提取超快去激发的结构动力学集体模式和轨迹。第二,结合易于处理和准确的计算方法,可以确定通过锥形交叉点去激发所涉及的电子态的形貌。第三,该方法可用于确定控制由数千个原子组成的分子的去激发动力学的关键集体变量和边界条件。第四,数据驱动的机器学习与现有的实验和理论技术相结合,提供了光谱的时间分辨率和结构方法的空间分辨率。
从五个实验动态轨迹推导出的圆锥形交叉点的地形和 PYP 中相关的人口动态。
在 PYP 的说明性示例中,研究人员的方法提供了以下新见解:
1、PYP 的反式到顺式异构化过程中涉及高频电荷振荡。这些振荡涉及先前忽略的发色团「外围」区域。
2、PYP 中上述振荡的存在已在 3-30 THz 的光谱可访问范围内得到独立证实,但没有直接的结构信息。研究人员的工作将实验可访问范围扩展到 100 THz,并提供有关这些超快振荡中涉及的结构元素的高分辨率空间信息。
3、光激发 PYP 通过五个管道之一向锥形交叉口前进,揭示了接近锥形交叉口的结构动力学轨迹。
4、通过锥形交叉口的结构-动力轨迹已经确定,可以测量 PYP 中锥形交叉口的关键参数。这些参数对于定量理解 PYP 中的 PES 流形和异构化过程至关重要。
5、研究人员的工作在结构和光谱研究之间架起了重要的桥梁。此链接对于全面了解 PYP 至关重要。
上述见解代表了对 PYP 以及广泛的「超快」结构动力系统的理解的明显进步。
实验数据是在 PYP 的时间分辨(光泵、X 射线探针)系列飞秒晶体学研究中获得的。已知这种蛋白质通过锥形交叉点进行快速的反式到顺式异构化反应。数据由二维(2D)衍射快照的时间序列组成,每个快照都来自通过三维(3D)衍射体积的不同随机中心切片。
由于光泵和 X 射线探针脉冲之间不可避免的「定时抖动」,在已知精度为 100 fs 的时间点进行光激发后记录每个「光」2D 快照。此外,在没有任何光激发的情况下记录了「黑暗」快照。
通常,来自同一标称时间点的足够多的光 2D 快照被索引和组合(合并)以获得 3D 衍射体积,并从那里获得每个时间点明暗原子结构之间的差异。时序抖动将合并的 3D 体积的时间分辨率限制为 100 fs。
论文链接:https://www.nature.com/articles/s41586-021-04050-9
相关报道:https://phys.org/news/2021-11-quantum-physics-proteins-ai-unprecedented.html内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢