基于测序的 RNA 结构探测可以生成 RNA 二级结构的全转录组谱。需要足够的结构覆盖才能获得关于 RNA 结构和功能的客观见解,但探测方法通常会产生不均匀的覆盖,在许多转录本中缺少结构分数。
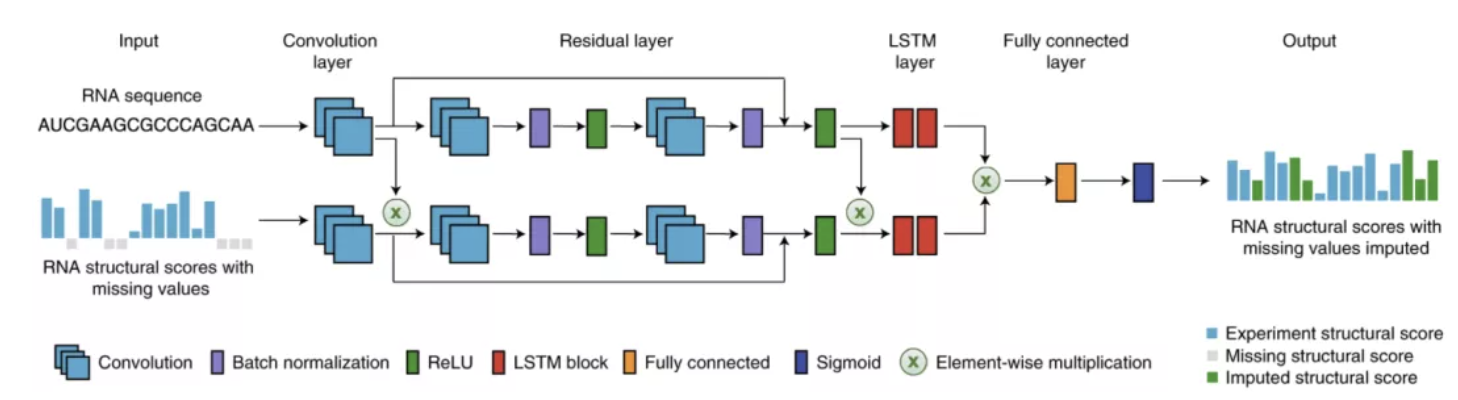
为了克服这一障碍,清华大学的研究人员开发了 StructureImpute,这是一种受计算机视觉深度补全启发的深度学习框架,它将 RNA 序列与相邻核苷酸的可用 RNA 结构信息相结合,以推断缺失的结构分数。
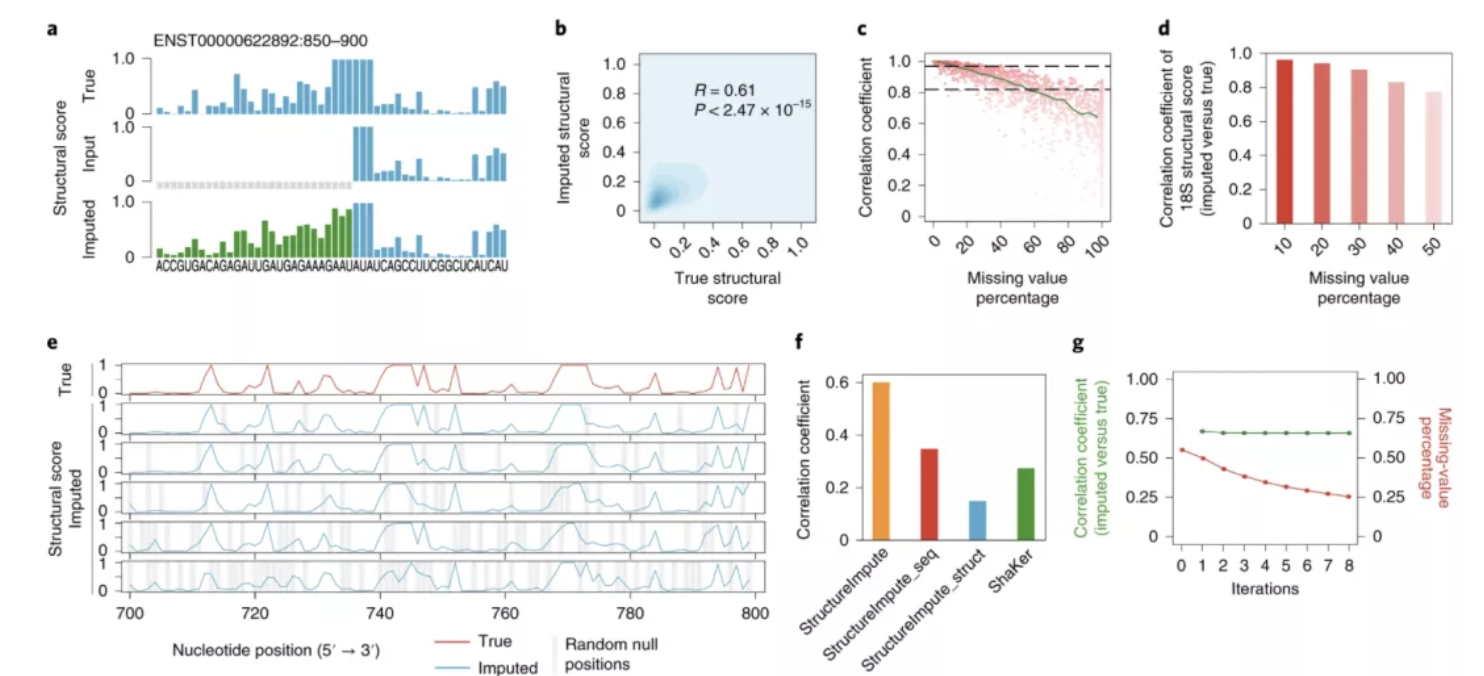
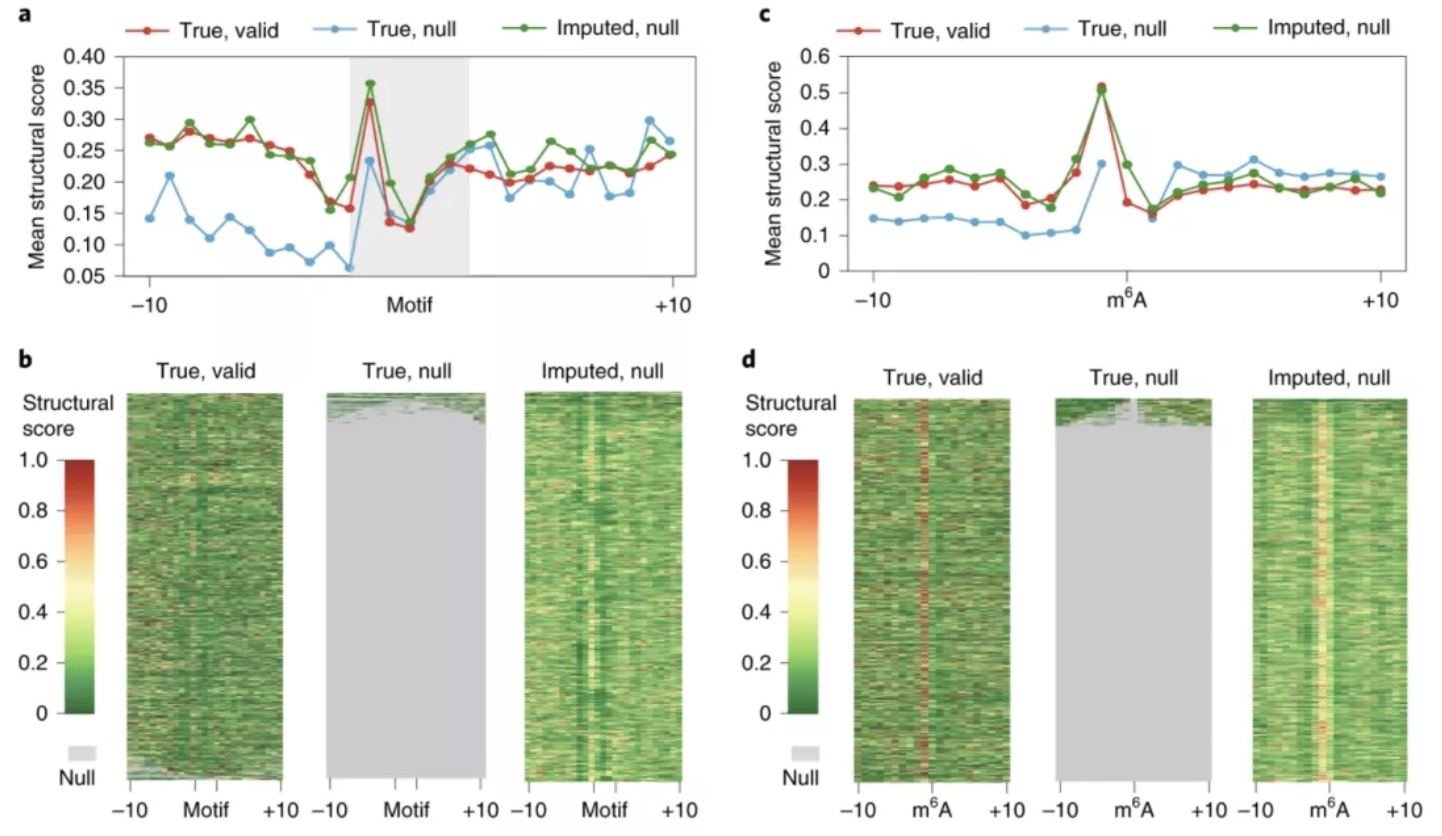
研究人员展示了 StructureImpute 强大的插补性能,其准确性远远优于仅基于 RNA 序列的预测。结果表明,StructureImpute 能够可靠地重建具有生物学影响的 RNA 调控区域的 RNA 结构模式,包括蛋白质结合和 RNA 修饰位点。最引人注目的是,StructureImpute 可以使用迁移学习来应用在一个数据集上训练的模型,以准确推断其他数据集中缺失的结构分数,即使它们是使用不同技术(例如,icSHAPE 和 DMS-seq)生成的。
该研究以「A deep learning method for recovering missing signals in transcriptome-wide RNA structure profiles from probing experiments」为题,于 2021 年 11 月 16 日发布在《Nature Machine Intelligence》。
转录本的非编码甚至编码区采用不同的 RNA 结构,这些结构在不同的生物过程中调节基因表达和功能,异常的 RNA 折叠通常会导致人类疾病。最近,已经开发了一套基于酶促或化学探测和深度测序的组合来绘制 RNA 二级结构的方法。这些方法利用 RNA 酶或小分子在特定结构环境中切割或修饰 RNA 核苷酸,然后使用深度测序定量检测逆转录(RT)停止或突变时的修饰。这些基于测序的 RNA 结构探测方法能够在转录组范围内检测 RNA 转录物的结构构象,从而彻底改变了 RNA 结构和功能的研究。
然而,尽管这些 RNA 结构探测方法有可能揭示整个转录组(RNA 结构组)的二级结构图谱,但获得的结构组的全面性通常还远未完成。结构覆盖取决于转录本丰度、特定核苷酸对特定探测试剂的可及性,以及测序文库构建过程中 RNA/互补 DNA (cDNA)片段的检索效率。例如,在结构探测中经常会遗漏低丰度的转录本(包括许多长的非编码 RNA)。
此外,即使对于丰富的 RNA,转录物的结构特征也常常是不完整的(即,包含许多核苷酸的缺失结构信号)。这种有限的结构覆盖可能会阻碍甚至使功能研究产生偏差,因为它会阻止检测具有功能影响的 RNA 结构。增加测序深度可以提高结构覆盖率;然而,与单细胞 RNA 测序(RNA-seq)研究面临的困境类似,这会成倍增加测序成本。因此,获得更完整的转录组范围 RNA 结构图谱的成本效益策略是有必要的,并将极大地促进 RNA 结构图谱研究在生物医学研究中的效用。
长期以来,RNA 序列本身一直被用于通过应用各种计算方法来预测结构信息,包括基于最小自由能假设、共同进化约束和现有结构知识的方法。然而,特定细胞环境中给定转录物的体内 RNA 结构是由其序列和细胞环境的组合决定的,包括例如反式因子,如 RNA 结合蛋白。
正如对不同细胞类型、亚细胞条件和状态的 RNA 结构探测研究所揭示的那样,这些反式因子在不同的不同细胞条件下会发生变化,并且可以显着改变 RNA 结构。因此,仅使用 RNA 序列的计算预测无法捕捉 RNA 结构的上下文依赖性,也无法反映不同生物条件下的结构变化,这对于揭示功能决定因素通常至关重要。
研究人员意识到,在某些方面获得更完整的结构图谱的目标类似于现在流行的插补策略,用于在单细胞 RNA-seq(scRNA-seq)研究中获得缺失的基因表达值(例如,作为 scImpute、MAGIC、SAVER 和 SCALE)。
事实上,类似于不同基因的表达相互依赖,这是 scRNA-seq 分析中基因表达插补的基础,相邻核苷酸的 RNA 结构也具有内在的相互影响。因此,研究人员建议利用来自相邻核苷酸的结构信息,只要可用,就可以估算 RNA 结构分析数据中缺失的结构分数。然而,与 scRNA-seq 基因表达插补任务相比,这里必须注意一个重要的区别:还应整合对基础序列的结构依赖性,以支持关于 RNA 结构组的更准确和生理相关的插补。
因此,研究人员没有从 scRNA-seq 插补中汲取灵感,而是追求 RNA 结构体插补在概念上更接近于自动驾驶中的计算机视觉挑战,称为深度完成,其目的是从物理获取的稀疏雷达数据和单目二维图像中恢复密集深度图。一种新开发的名为 GuideNet 的深度学习方法是一种用于深度完成信息集成的最先进的神经网络架构,它使用成对的红绿蓝(RGB)图像作为卷积滤波器来引导光检测和测距点云完成。GuideNet 在多个数据集上的表现明显优于其他完成方法。
受 GuideNet 的启发,研究人员开发了一种名为 StructureImpute 的深度学习模型,该模型通过将 RNA-seq 数据与来自这些分析实验(例如,使用 icSHAPE 或 DMS-seq)的可用 RNA 结构数据相结合,对从基于测序的 RNA 结构分析实验中获得的结构组中缺失的结构信号进行估算。
图示:用于 RNA 结构评分插补的 StructureImpute 的整体架构。(来源:论文)
通过在各种数据集上测试 StructureImpute,既证明了 StructureImpute 作为增强 RNA 结构分析数据集的方法的有效性,又说明了 StructureImpute 在准确推断转录组中具有生物学影响的功能位点的缺失结构信号方面的效用。
令人兴奋的是,它提供了一个生物学示例,说明从丰富数据集上训练的模型中进行迁移学习如何支持从稀疏数据中进行准确预测;研究证明,从体内 HEK293 全细胞数据集训练的 StructureImpute 模型可以对亚细胞组分、体外数据集甚至 K562 细胞和成纤维细胞的数据集中的 RNA 分子进行准确和生物学信息的结构预测。
在过去的十年中,已经开发了许多基于测序的方法来揭示转录组范围内的 RNA 结构景观。然而,它们的应用受到各个位置的低结构覆盖率的限制。为了克服这一障碍,研究人员建立了一种新的深度学习方法 StructureImpute,它可以估算缺失的 RNA 结构分数,从而增加转录组的结构覆盖率。基于精心设计的网络模块,包括 LSTM 模块,StructureImpute 推断嵌入在初级序列中的结构信息,并学习相邻核苷酸的结构信息的内在依赖性。序列和相邻结构约束(即从探测实验中获得的部分结构轮廓)的这种整合使得比仅序列方法具有更好的插补性能,该研究证明了这一点适用于不同的数据类型和多种应用。
图示:StructureImpute 的性能评估。(来源:论文)
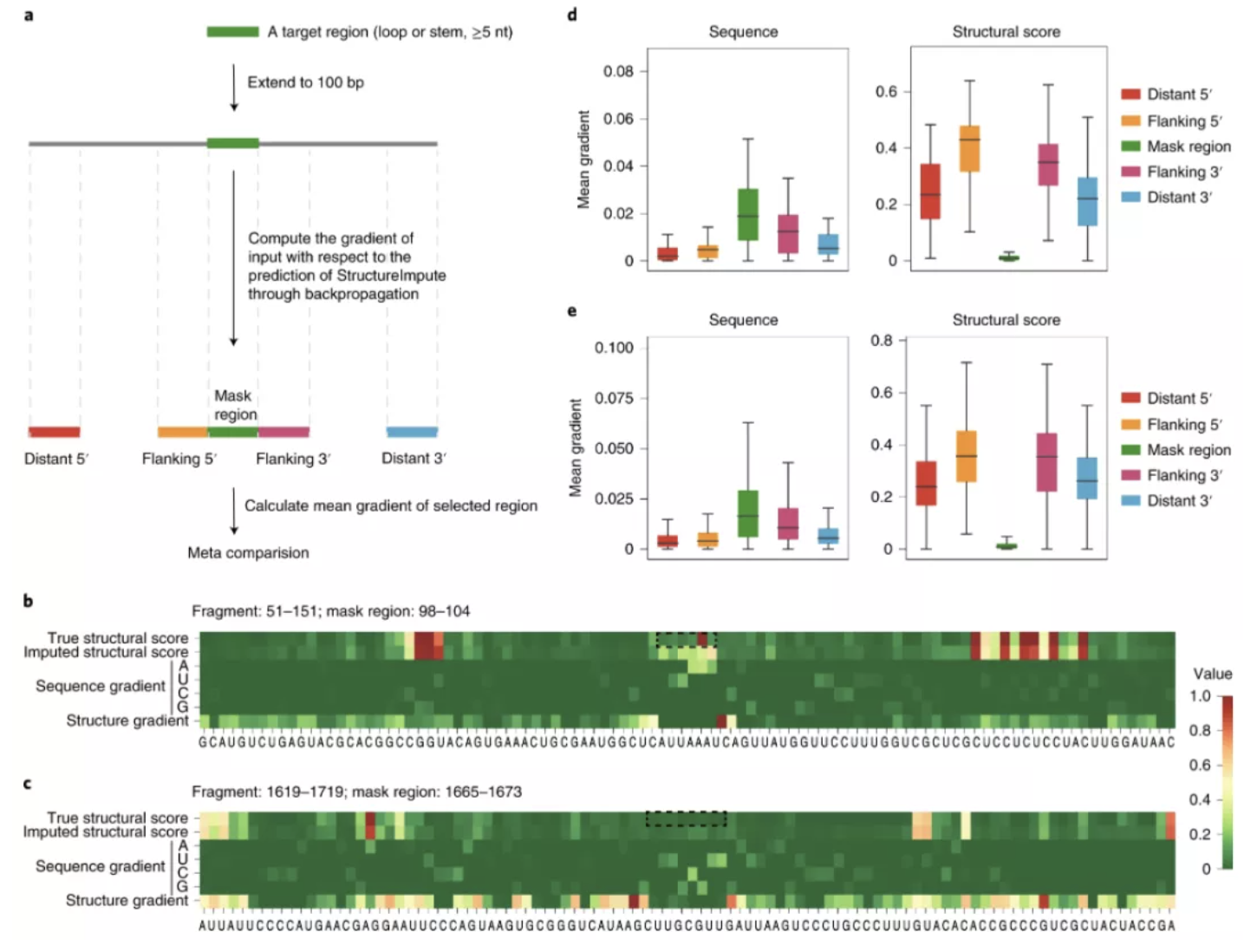
生物环境之间的这种高度准确的性能甚至不同的 RNA 结构分析方法令人有些惊讶,研究人员推测 StructureImpute 在这些非常多样化的生物样本中的强大性能与 RNA 分子固有的一些因素有关。也就是说,RNA 转录本的结构已被广泛认为取决于其 RNA 序列和特定的细胞环境;同时,给定核苷酸的结构直接受其侧翼核苷酸的影响,有时还受结构特征和远端区域存在的核苷酸的影响。
因此,直接和间接的结构相互作用共同决定了 RNA 分子的结构。为了获得侧翼和远处区域的好处,研究人员应用了一个 LSTM 块,该块已被证明能够记住长期依赖关系。研究人员推测最初训练的 StructureImpute 模型,学习了如何利用相邻核苷酸之间甚至可能是远端核苷酸之间的内在结构依赖性,并且可能输出反映这些结构依赖性的预测。
图示:RNA序列和结构信息对StructureImpute插补性能贡献的梯度分析。(来源:论文)
因此,StructureImpute 结合了序列和实验结构信息来估算 RNA 结构分析实验中缺失的结构信号,并且它可以很好地完成这项任务。但是,未来的努力将集中在解决当前的一些限制上。
首先,当前的 StructureImpute 模型是使用相同的、相对较短的 RNA 片段(此处为 100 nt)进行训练的,因此给定转录本的数据通常必须拆分成箱进行插补。拆分转录本不可避免地会导致超出所采用长度限制的结构依赖性丢失,例如 5' 和 3' 非翻译区域之间的潜在相互作用。
图示:StructureImpute 准确地估算功能区域内缺失的结构分数。(来源:论文)
研究人员用 400 nt、200 nt 和 100 nt 的不同片段长度测试了不同模型的训练。从 100 nt 开始,因为之前的研究表明大多数局部碱基配对都在这个距离内,但也有很多局部碱基配对长于 50 nt。研究人员计算了所有模型的真实结构分数和预测结构分数之间的相关性。研究人员发现模型性能随着片段长度的增加而下降,这表明使用更大片段的建模不一定会提高性能。
最初的假设是,在较大片段的输入配置文件上训练的模型将有助于捕获长距离碱基配对。然而,实际上观察到了插补性能的相反趋势。研究人员推测长距离碱基配对可能相对更难捕获,可能需要更复杂的模型。因此,值得探索支持更长片段甚至整个转录本的插补的框架。
其次,研究人员注意到插补精度随着缺失结构信号水平的增加而下降。这一发现既证实了先前报告的仅序列预测方法的局限性,也强调了来自相邻核苷酸的结构约束对估算准确结构分数的重要贡献。然而,丢失信号的长片段在实践中非常普遍。更好的深度学习模型和/或更多的训练数据应该有助于解决这个问题。
具体来说,设想 StructureImpute 的主要用途是将其应用于结构组以增加结构分数的覆盖范围。在这里采用了分箱 + 迭代方法,该方法在可能的情况下在每次迭代中对每个窗口(或分箱)中的分数进行估算(例如,对于窗口中缺少的值少于 50% 的情况)。一旦估算了一些分数,研究人员将估算的分数作为输入的一部分,以在下一次迭代中进一步估算剩余的分数。使用滑动窗口策略在每个滑动窗口中插补一个分数,然后加入预测分数以最大化插补结构分数的覆盖范围将是一个合适的替代方案。
第三,StructureImpute 实现了一个端到端的模型框架。这种方法可以有效地训练网络,但限制了识别哪些输入特征和设计的模块影响插补性能的能力。这些信息对于进一步改进该方法非常重要,并且应该有助于指导其在日益多样化的数据集上的适当应用。
最后,这里应该考虑深度学习研究中常见的一个重要趋势:已知用于训练模型的一个数据集的丰富性可以显着改善模型在其他包含稀疏信息的数据集上的应用。
这种迁移学习显然是 StructureImpute 的一个吸引人的属性,值得强调:该团队可以在具有非常深的测序深度的高质量数据集(例如,人类 HEK293 细胞的基准数据集)上训练 StructureImpute 模型,然后应用该模型对另一个测序深度低得多的数据集进行插补。
因此,StructureImpute 允许所有研究人员从最高质量的可用 RNA 结构数据集(这些数据集在生物学和医学的各个领域出现时在技术上仍然非常困难且成本高昂)中获益。
探索用于模型训练的序列和结构覆盖数据的丰富性如何最终影响模型,对各种稀疏应用程序数据集的插补性能将非常有趣。这样的研究可以产生生物信息学、深度学习甚至生物学的新发现。
论文链接:https://www.nature.com/articles/s42256-021-00412-0#Sec1内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢