
【论文标题】fastMSA: Accelerating Multiple Sequence Alignment with Dense Retrieval on Protein Language
【作者团队】Liang Hong, Siqi Sun, Liangzhen Zheng, Qingxiong Tan, Yu Li
【发表时间】2021/12/21
【机 构】港中文、微软、深圳先研院等
【论文链接】https://doi.org/10.1101/2021.12.20.473431
进化相关的序列为蛋白质的结构和功能提供了信息。多序列比对,包括从大型数据库中搜索同源物和序列比对,可以有效地挖掘信息,帮助蛋白质结构和功能的预测,其效率已被AlphaFold证明。尽管已经有了多序列比对的工具,但从整个UniProt中搜索同源物仍然是很耗时的。考虑到AlphaFold的成功,可以预见,针对大规模数据库的大规模多序列比对将成为该领域的一个趋势,加快这一步骤是非常必要的。本文提出了一种新的方法,即fastMSA,以显著提高速度,该想法与以前所有的加速方法是正交的。本文提出了一个新颖的双编码器架构,利用基于BERT的蛋白质语言模型,可以将蛋白质序列嵌入到一个低维空间,并在运行BLAST之前有效地过滤不相关的序列。广泛的实验结果表明,本文可以以34倍的速度召回大部分的同源物。此外,本文的方法与下游任务兼容,如使用AlphaFold进行结构预测。使用本文的方法生成的多序列比对,本文在蛋白质结构预测方面的性能几乎没有受到影响,而且运行时间更短。fastMSA将有效地帮助基于同源物和多序列比对的蛋白质序列、结构和功能分析。
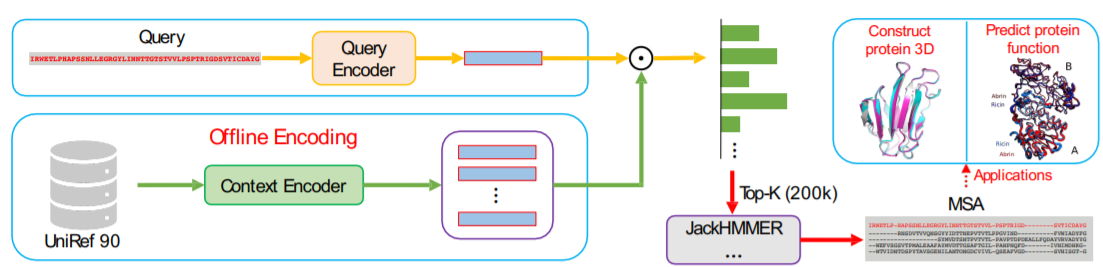
上图展示了fastMSA的框架在,它由两个基于Transformer的模型组成,作为查询序列编码器和候选序列(来自非常大的数据库的序列)编码器。使用点积检索出前K个最相似的序列,然后将JackHMMER应用于这个小的检索数据集,为进一步的任务建立MSA,如三维结构预测或蛋白质功能预测。在检索之前,UniRef90可以被离线编码为向量,这不会影响构建MSA的推理时间。
- 从原始序列中构建训练数据,使用JackHMMER v3.3从UniRef90中搜索查询序列,并建立MSA作为真实标签。
- 从Transformer编码器选择第一个向量作为其序列表示,学习序列嵌入后计算这些序列嵌入之间的内积
- 按照对比学习的方法,进一步使用批内数据作为底片。这与底片的实际抽样相比,这可以显著提高训练效率。更具体地说,对于一批成对的数据(q1,c1), .... ,(qb, cb),其中b是批次大小,查询qi的负面样本是同一批次内的所有其他候选数据,任务是在所有其他否定的候选者cj中确定同源物ci,这是一个b分类问题。
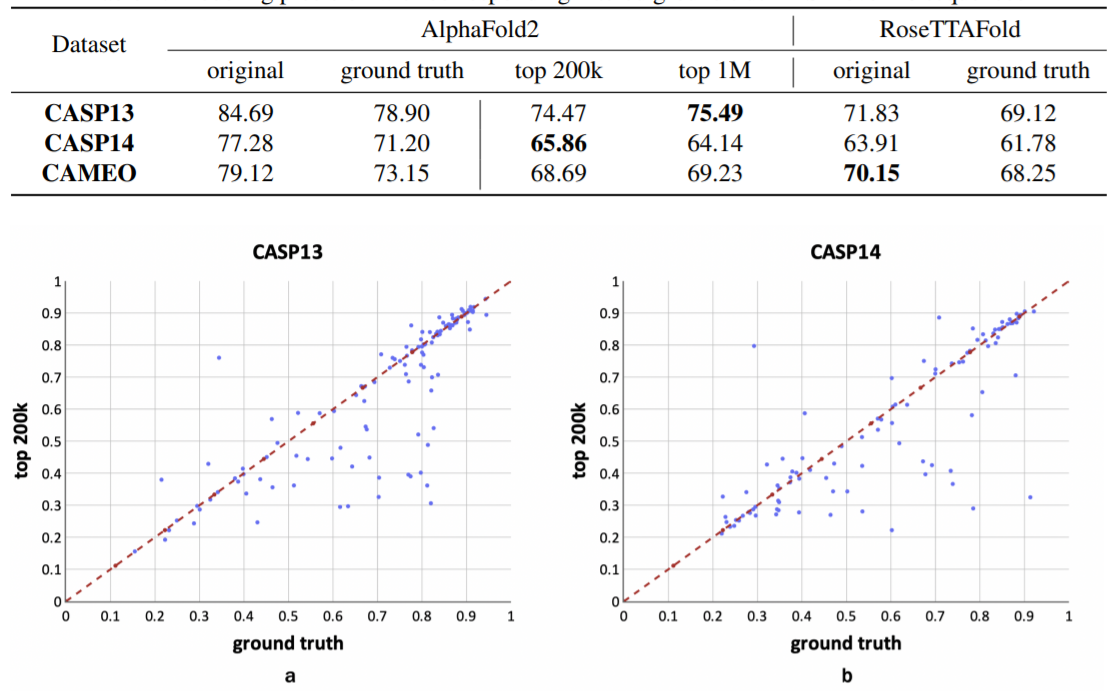 上图为使用真实的MSA和本文的方法得到的MSA对每种蛋白质的三维结构建模精度进行比较。本文使用AlphaFold2来预测MSA的三维结构。 a图表示CASP13蛋白的真实MSA和fastMSA的MSA经过top-200k过滤后的三维结构建模精度的头对头比较。b图展示CASP14蛋白的真实MSA和fastMSA的MSA经过top-200k过滤后的三维结构建模精度的头对头比较。可以发现在三维结构建模中,来自fastMSA的MSA可以与真实MSA相媲美,有时甚至优于后者。
上图为使用真实的MSA和本文的方法得到的MSA对每种蛋白质的三维结构建模精度进行比较。本文使用AlphaFold2来预测MSA的三维结构。 a图表示CASP13蛋白的真实MSA和fastMSA的MSA经过top-200k过滤后的三维结构建模精度的头对头比较。b图展示CASP14蛋白的真实MSA和fastMSA的MSA经过top-200k过滤后的三维结构建模精度的头对头比较。可以发现在三维结构建模中,来自fastMSA的MSA可以与真实MSA相媲美,有时甚至优于后者。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢