本文是一系列文章的整理,包括一些本公众号以前推送过的,也包括一些最新发表还没有经过同行评议的以机器学习辅助的蛋白酶改造方法为主题的文章。希望能够梳理一下(片面的)随着机器学习方法的大跃进,在酶工程领域计算方法有哪些进展,也希望能得到读者的补充、批评和指正。
第一部分 传统方法
这里所谓的传统方法主要指的是 2008 年前后,由于 Rosetta 软件对蛋白质建模的能力取得突破,一系列自然界中没有的酶(尚未发现具有该功能的酶)被设计出来[1-3],需要与蛋白质从头设计(de novo design)加以区别的是,de novo enzyme design 强调的是设计酶的活性中心是全新的,而不是简单的从另一个相似功能的酶上面嫁接(graft)过来。全新的活性中心一般被放置在蛋白骨架(scaffold)上,并通过设计使其稳定。这一系列方法在[4]中有非常具体的介绍。
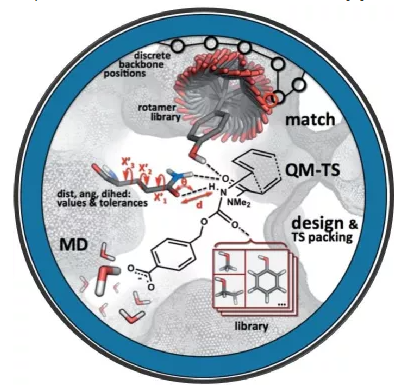
基于结构建模的方法
在一个蛋白上重新改造活性中心使其具有完全不同于以往的活性无疑是困难的,前述设计的酶的活性也比自然界进化出的高效酶差很多。另一个相对简单的酶改造的思路是保留酶原来的活性中心,通过对底物结合口袋的改造来调节底物特异性,扩展或改变酶对底物的选择性。
吴边等的一系列工作[5,6]展示出,基于结构建模的方法以及对化学反应的机制的理解,通过在活性中心设计时引入大量先验的约束条件,可以以较高的成功率设计得到目标底物的酶突变体,甚至可以实现对手性的控制。
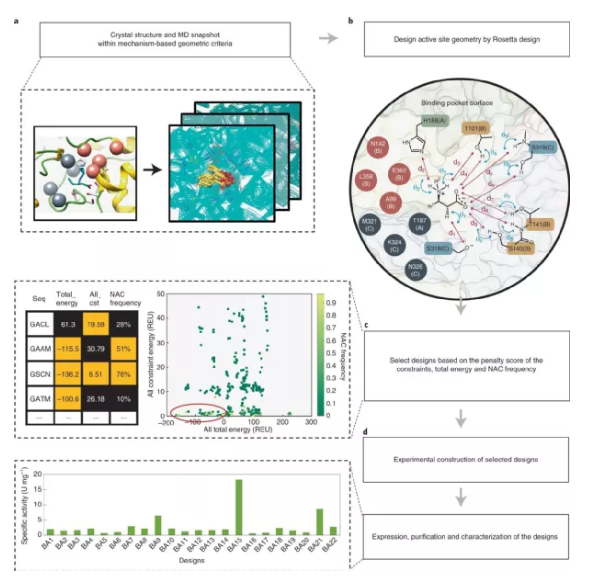
基本概念
下面简单介绍一下 David Baker 组开创的 Rosetta 软件中进行蛋白质建模的一些基本概念和主要方法。分子建模问题中最重要的两个概念是采样和打分。Rosetta 的打分函数除了从charmm 力场中搬来的具有物理意义的原子相互作用(vdw,elec 等)还加入了大量的基于统计的(knowledge-based)打分项并且各项之间的权重会根据不同的应用场景进行优化,使它在不同的 benchmark 中取得较好的表现。
侧链及其不同的构象被称作 rotamer,自由度在内坐标空间仅允许 chi 角进行转动(如果需要也可以进行笛卡尔空间的采样)。在蛋白结构优化时,每个侧链只采样同一类型氨基酸的rotamer,而设计时可以采样用户指定的氨基酸集合的 rotamer, Rosetta 中的杀手应用packer 可以高效的处理这种排列组合问题。但由于采样空间被离散化成有限的 rotamer 的组合,采样不充分,打分也需要相应调整,在 pack 之后再进行梯度下降的优化微调结构,多次重复这一过程(relax),可以让结构高效的进入能量最低状态。
软件中有多种采样(mover)和打分方法(score)可供选择,一个成功完整的设计流程需要这些元素的合理组合,这与初始结构的质量,设计的目标等都有关。采样方法会有偏倚, 打分函数也会有漏洞,并不是计算得到的打分越好结果成功率越高。原则上,尽可能控制采样空间的大小,引入生信提供的约束条件,采用量化计算的结构约束,都可以提高最终设计的成功率。
如果对更多的内容感兴趣,可以参考几篇知乎上的文章[7,8],这里有一些基本原理的更具体介绍,以及一个比较近的通用设计策略 Funclib 的介绍。
基于数据库的方法
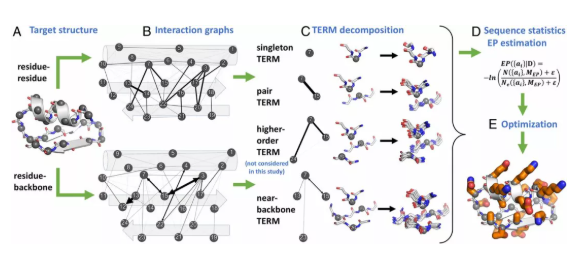
另一类蛋白建模策略则是完全基于数据库(PDB)的分析。Gevorg Grigoryan 等开发了一系列工具可以对 PDB 结构进行搜索(MASTER[9])和 motif 提取(TERM[10,11]),并利用这些工具对 PDB 数据集进行分析,得到了各种 motif 的分布,并以此为打分可以实现对蛋白质结构的评估甚至设计(dTERMen[12])。
![]()
定向进化方法
当然计算设计并不是对酶进行改造的主流方法,通过“突变-筛选”这一师法自然的定向进化方案仍然是最广泛使用的对酶进行改造的策略,Frances Arnold 也因为在该领域的重要贡献而得到 2018 年诺奖。该方法得到的酶的活性显著优于计算设计结果,一般计算设计得到一个“起点”酶之后,往往也会接一步定向进化。从这个意义上说,两个方法其实是互补关系,计算可以在较大的序列空间中进行探索,缺点是精度不够,而定向进化则是在局部构象空间中进行准确的微调。当然两个方法也在分别寻求突破,计算希望能够提高预测精度,而定向进化希望增加突变体的覆盖空间和采样效率,后面章节会介绍“机器学习”成为两种策略不约而同的选择。
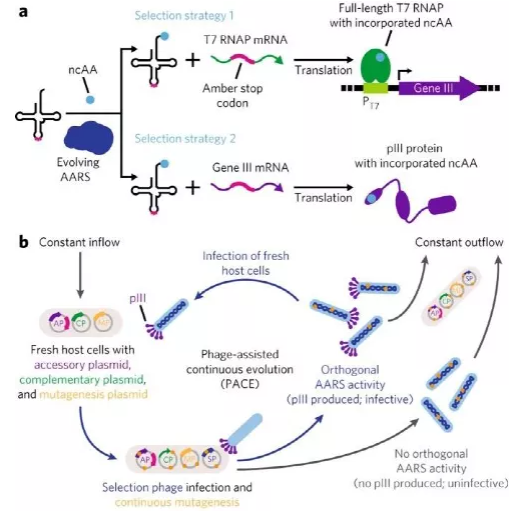
定向进化是一个强有力的工具,可以利用在选择压力下反复诱导复制过程中的突变,生成具有目标活性的蛋白。然而监控这一过程中的全长序列及其频率的演化轨迹则非常困难,大多数高通量DNA测序方法,很难得到全长基因序列、频率以及适应度(fitness),因为短读序列可能会丢掉单倍型中突变的连接片段。后文会介绍机器学习如何帮助解决这一问题。
第二部分 经典机器学习
机器学习方法一直在各个领域帮助人们解决从数据中找规律的问题。与基于物理的方法不同,它不要求总结出来的规律具有明确物理意义,只有一个宗旨就是好用就行。机器学习也经历过寒冬,但随着算法的不断演进以及计算机性能的指数增长,目前几乎在各个领域,都需要用到机器学习来帮助改进本领域的方法。一篇 2020 年的综述文章介绍了机器学习在酶工程的应用[15]。
机器学习方法辅助建库
Frances Arnold 组从 2019 年就报导了机器学习对定向进化建库的帮助[16,17]。他们发现通过机器学习算法对随机突变文库的筛选结果进行学习,可以加速定向进化对酶活性的优化。今年他们又发表了一篇综述文章[18],更深入的讨论了最新的深度学习方法在这个领域的应用,我们会在下一章展开介绍。
定向进化过程的一个时间点可能包含巨大的基因多样性( >10^7),尽管单个突变可以通过鸟枪测序和基于交叠图的装配进行测序,这些方案很难推广到定向进化产生的多样性规模。一般人们采用 Illumia 二代测序技术,但其读长往往远比基因全长要短,这使得同一基因中的远距离突变关联被破坏。高通量长读长的方法目前还不够完善,错误率也较高。David Liu 于近期开发了Evoracle[19],可以使用定向进化实验不同时间点的短读数据,精确地重建全长基因序列及其适应度轨迹,比同类方法有显著改进。该方法使用包含噪音的短读数据推理非线性动力学模型中的隐含参数。他们在三种体系中验证了Evoracle 方法,包括:对腺嘌呤碱基编辑器的 PACE 和 PANCE 进化,以及对抗药酶的 OrthoRep 进化。Evoracle 方法显著优于两种 overlap-free 的重建方法,并在相邻核苷酸之间连接片段完全丢失且测量噪声大的数据(如pooled Sanger 测序数据)上,Evoracle 保持了强劲的性能(R^2 = 0.86)。进一步研究表明,Evoracle 还可以远远早于保守性分析识别出高适应度变体,包括低频“rising star”,大大提高了关于基因变异适应度的训练机器学习模型的可行性。
结构方法与机器学习方法的结合
一篇最近发表的Cell 文章[20]展示了建模方法与定向进化方法的成功结合。文章首先采用传统方法进行底物特异性设计,在得到了具有一定活性的“起点”酶后,设计实验进行了两轮定向进化实验。首先使用 Rosetta 进行结合口袋的设计,接着在口袋区域进行单位点饱和突变并将实验结果用随机森林方法进行训练定位四个重要位点,最终对这四个位点进行组合突变并采用广义线性回归方法建模,并经过多轮预测筛选,得到了性能非常好的突变体。具体内容可以参考我们之前的Cell | 机器学习指导血清素传感器的定向进化。
第三部分 深度学习
下面进入本文的重点部分,也就是深度学习技术如何取得突破性进展并以一种意料之外但又情理之中的方式深刻影响蛋白质研究。
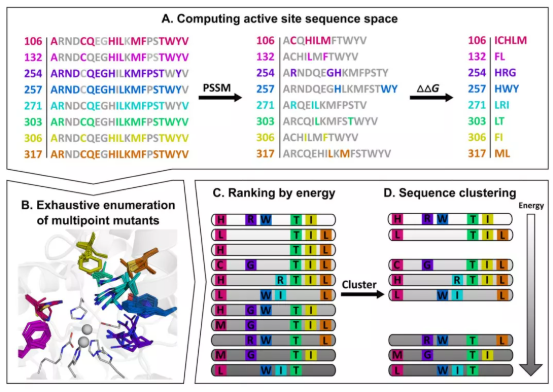
早期生物信息学家利用序列比对等工具研究蛋白质家族的序列保守性如 PSSM(position-specific scoring matrix),可以注释蛋白功能,定位活性位点等。这时所使用的是单独氨基酸位点的独立信息。很快人们认识到,蛋白质序列的进化(或者说演化可能更加准确)过程,位点之间的突变并不是完全独立,位置靠近或者功能相关的(往往位置也不会远) 位点之间突变存在相关性。单纯的相关性分析或者互信息分析可以提供一部分信息,但由于相关性可以传递,并不能据此来很好的预测残基间的接触(contact)。近十年来,涌现出一系列新的算法如EVfold[21],plmDCA[22],Gremlin[23]等,采用不同的统计物理技术,试图引入一些简化计算残基对之间以及残基和全局之间的关系。近期的一篇文章发现这些方法也都可以放到一个统一的深度学习自编码器框架下来理解[24]。
而深度学习的一大突破来自于计算机图形学(CV),随着 CNN 和 Resnet 等技术的发明和广泛使用,机器学习在图像识别,图像语义分割等领域的性能达到甚至超过人类水平。另一大突破来自于自然语言(NLP)领域,随着RNN 替代 HMM,Transformer 又替代 RNN,机器学习在文本分类,语义分析,信息提取,语音识别,语言翻译等领域的表现也逐渐赶超人类水平。那么这些进展和蛋白质有什么关系呢?
非监督的深度学习方法
一个最直接的类比就是,基因序列其实就是文本,是自然创造生命的“天书”,是真正的“自然语言”。套用 NLP 领域的预训练(如 BERT)方法,可以对基因组计划以来产生的海量原始基因组数据(蛋白质序列)进行建模,从而学习到氨基酸编码蛋白质的内在的规律。再应用到下游的具体任务。
George Church 等开发的 UniRep 模型[25],基于双向 LSTM,在 UniRef50 的序列数据库上(约 24 million)进行预训练,得到的模型可以把任意序列的每个氨基酸编码为一个上千维度的向量,近而用于其它分类、预测问题。
Facebook 开发的ESM 模型[26],基于 Transformer,在 250M 大小的UniParc 数据库上进行训练,得到了 SOTA 的结果,他们还进一步开发了基于 MSA 的和基于 fitness 的ESM-MSA-1b 以及 ESM-1v 版本(后面说)。
有监督的深度学习方法
另一个类比由许锦波等首先意识到[27,28],蛋白质三维结构可以表示成为一个二维的接触图谱(contact map),而这个图的绘制则类似于 CV 中的语义分割问题,输入的蛋白质序列如果可以整理成多通道的二维矩阵,则可以把它当做一个图片来处理。他们首先在CASP 的contact 预测比赛中取得佳绩,随后启发了 DeepMind 开发出AlphaFold[29]在随后的一届CASP 正赛中夺冠。他们的主要贡献是证明深度学习架构可以准确预测残基间的距离分布,将分布转变为能量约束后,可以通过简单的能量最小化得到非常好的效果。学界随后开发出trRosetta,除了预测距离分布,还增加了键角二面角等刚体相关约束,并用更少的计算资源反超 AlphaFold,更具体的介绍可以参考trRosetta: 在台式机上从头折叠一个蛋白——这台台式机最好有一块好显卡。
去年的CASP 竞赛中AlphaFold2 横空出世,用全新的架构将蛋白质结构预测的精度推到了前所未有的水平,已经在很多目标中达到了实验级别。学界根据DeepMind 开会时介绍的思路,开发出 RoseTTAFold,接近 AlphaFold2 的精度,并在蛋白复合物预测中取得进展。近期谷歌开源了自己的算法,也发布了对重要物种的全蛋白质组结构预测数据集,还公开了蛋白复合体预测新模型AlphaFold-Multi。这些新闻最近已经被报道得很多,就不赘述了。
但这里想介绍一些在线资源,其实现在任何人想进行这些预测只要能科学上网,就可以使用谷歌的免费Colab 服务进行。首先谷歌自己就推出了一个在线预测版本[30],只需要输入序列就可以进行单体和复合物的预测。ColabFold[31]是 Sergey Ovchinnikov 在 AF2 公布的代码基础上进行魔改,提高了 MSA 构建速度,同时还增加很多复杂的复合物构造策略,可以进行非常 fancy 的结构预测。这些操作都可以在网页上简单操作就可以完成。
生成方法
前面介绍的深度学习的进展主要都是在结构预测的领域,下面我们回到本文的主题,如何改造酶。改造,就是要产生满足要求的新序列,所以我们还需要介绍一下蛋白序列的生成模型。这一领域其实也有多个工作发表,这里挂一漏万的简要介绍一下。
DeepSequence[32]是 Debora S. Marks 在 2018 就发表的一篇文章,利用变分自编码器(VAE)的思想,通过隐变量模型建模每个氨基酸到全局的关系,文章展示这个模型对点突变的预测能力好于单体和两体模型(如他们自己 2017 年发表的 EVMutation)。并建立了一套评估序列生成模型好坏的测试集,对随后的研究工作有重大影响。
ProGen[33]于 2020 年上线,在大约 2.8 亿的蛋白序列上训练了巨大的语言模型(1.2B 参数),文章展示通过非监督学习预训练的方式得到的大规模语言模型并没有发生过拟合,模型的性能随着规模可以进一步提升,具体介绍可以参考这篇 ProGen:蛋白质生成语言模型 (https://zhuanlan.zhihu.com/p/250970069)。
ProteinGAN[35]是2021年发表的一个生成模型,利用生成对抗网络(generative adversarial network)的思想,本质上也是在学习蛋白序列的内在高阶分布的性质,并在一个模型蛋白上展示出将近 1/3 的生成序列具有目标活性。更详细介绍可以参考这篇 Nat.Mach.Intell | ProteinGAN:利用生成对抗网络扩展功能蛋白序列空间 (https://zhuanlan.zhihu.com/p/363295730)。
最后介绍一下 trDesign[36]。这是第一个把本来用于结构预测的监督模型逆转,用于序列生成的模型,之前的生成模型都是单纯基于序列,或者增加一些任务相关的标签,而 trDesign 方法是一个基于结构设计序列(也就是传统的 Design)的深度学习方法。而另一篇刚发表于nature 的“幻想”文章[37],则证明通过最大化 trRosetta 预测的分布与背景分布的差异,模型甚至可以凭空“想象”出一个合理的蛋白结构及其序列。更进一步,两篇还未正式发表的工作[38,39]展示,可以通过对模型的巧妙设计,在给定部分结构(motif)的情况下幻想蛋白的其它部分并设计序列,这为蛋白质的 binder 设计以及酶的改造提供了新的可能性。
更具体的介绍可以参考这篇 最终幻想: 无中生有的蛋白质从头设计 (https://zhuanlan.zhihu.com/p/185073475)和这篇 梦幻设计:使用深度学习设计功能蛋白质 (https://zhuanlan.zhihu.com/p/331821987)。
酶的改造方法
2020 年 7 月发表了一篇利用 bmDCA 模型进行序列采样优化酶活性的文章[34],这个方法并不属于深度学习,而是利用 DCA 方法基于多序列比对建立序列统计模型,并在此基础上进行序列采样,文章展示出在同源序列较多的情况下,学到的序列模型可以很好的反映出训练集中氨基酸的高阶统计性质,并生成出具有高活性的全新序列。我们也曾推送过这篇文章Science | 基于进化模型的酶设计。
UniRep 方法的一个在酶改造上的具体应用,low-N 方法[40],首先基于在 UniRef50 上的预训练模型 UniRep,再对所要研究的蛋白家族的同源序列集合进一步训练(eUniRep),利用该针对具体家族的模型对有监督的序列进行编码并进行简单的机器学习建模(evotuning)。文章展示这种优化的序列建模方式可以在很少的监督数据集(少于一百)中得到很好的拟合表现,暗示 eUniRep 可以很好的描述蛋白家族的序列特征。具体介绍可见Nat. Methods | 使用数据高效深度学习实现低N蛋白质设计。
ESM-1v[41]是 facebookESM 模型的针对突变影响的预测问题进行的升级,使用蛋白语言模型,作者展示即使没有任何标签,该模型仍然可以在 fitness 等任务中表现出色,实现所谓“zero-shot”的预测能力。作者发现使用 90%的序列数据库可以比之前的 50%库有很大提升,但 100%的数据库训练结果又会明显下降。
最后推荐一篇中文综述[42],以及一篇英文综述[43]。
评测方法
介绍了这么多方法,毕竟很难一一用实验验证,怎么判断哪个更靠谱呢?近期也有一些新的数据集构建和测评分析的文章发表出来。如 FLIP[44]和 MaveDB[45]。
第四部分 总结与展望
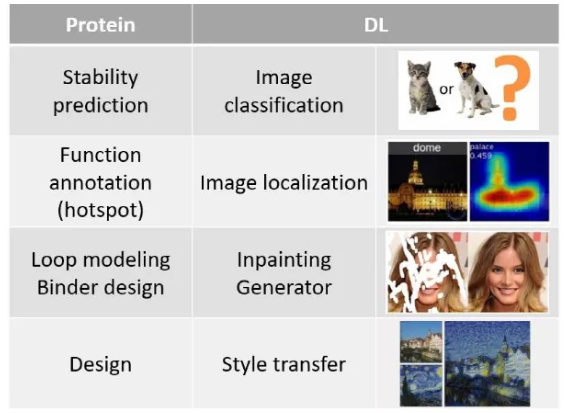
近几年来深度学习方法取得重大进展,尤其在图形学和自然语言问题中性能大幅提升。而很多这类问题在蛋白质建模问题中都有相似的结构。比如对蛋白质的稳定性预测可以类比图片分类问题,蛋白质的功能位点注释可以类比图片分割,loop 建模可以类比图片修复,蛋白设计(比如稳定性增加)可以类比风格迁移等。
而随着AF2 的横空出世,机器学习对蛋白质建模的精度达到了前所未有的高度,很多之前只是理论上可以work的工作已经可以走向实际。同时基因组数据以及蛋白质结构数据也在不断增加,实验技术的进步让更大的蛋白质复合物的数据集不断扩充,相信对蛋白蛋白相互作用的预测也很快能达到实验精度的水平。虽然现在蛋白质与小分子的建模还存在瓶颈,但图模型等新的网络架构仍在发展,人们对小分子的预测能力也在不断提高,希望不远的将来,人们可以针对任意感兴趣的位点设计小分子或蛋白药物。
当然作为一个物理问题,蛋白质折叠问题还没有被解决。整个动力学过程的物理规律人们还无法完全掌握,传统的分子动力学等方法依然起到重要的作用。近期Anton3 的发布让很多人惊掉下巴,似乎不远的将来人们就可以像看电影一样直接模拟蛋白质在较大时间尺度上运动规律。
如果说现在的机器学习方法像是“小无相功”一般可以作为任意任务的“内功”驱使各个门派的武功招式,基于基本物理规则的计算模拟就像玄铁剑“重剑无锋,大巧不工”。不知是否会有一天,人们可以统一机器学习与物理,贯通信息与能量,达到“以无剑胜有剑的化境”。
参考文献
1)Kemp elimination catalysts by computational enzymedesign
2)De Novo Computational Design of Retro-AldolEnzymes
3)Computational design of an enzyme catalyst for a stereoselective bimolecularDiels-Alderreaction
4)De Novo Enzyme Design UsingRosetta
5)Computational redesign of enzymes for regio- and enantioselectivehydroamination
6)Development of a versatile and efficient C–N lyase platform for asymmetric hydroamination viacomputational enzyme redesign
7)在 Rosetta 中计算氨基酸影响的方法
8)Funclib: 高效设计功能多样的酶库
9)Rapid search for tertiary fragments reveals protein sequence–structure relationships
10)Tertiary Structural Propensities Reveal Fundamental Sequence/StructureRelationships
11)Tertiary Alphabet for the Observable Protein StructuralUniverse
12)A general-purpose protein design framework based on mining sequence–structure relationships inknown proteinstructures
13)Data-driven computational proteindesign
14)定向进化技术的最新进展
15)Machine Learning in EnzymeEngineering
16)Machine learning-assisted directed protein evolution with combinatorial libraries
17)Machine-learning-guided directed evolution for protein engineering
18)Advances in machine learning for directedevolution
19)Reconstruction of evolving gene variants and fitness from short sequencing reads
20)Directed Evolution of a Selective and Sensitive Serotonin Sensor via Machine Learning
21)Protein 3D structure computed from evolutionary sequence variation
22)Fast pseudolikelihood maximization for direct-coupling analysis of protein structure from manyhomologous amino-acidsequences
23)Assessing the utility of coevolution-based residue–residue contact predictions in a sequence-andstructure-richera
24)Unified framework for modeling multivariate distributions in biologicalsequences
25)Unified rational protein engineering with sequence-based deep representationlearning
26)https://github.com/facebookresearch/esm
27)Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep LearningModel
28)Distance-based protein folding powered by deeplearning
29)Improved protein structure prediction using potentials from deeplearning
30)https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb
31)https://github.com/sokrypton/ColabFold/
32)Deep generative models of genetic variation capture the effects ofmutations
33)ProGen: Language Modeling for ProteinGeneration
34)An evolution-based model for designing chorismate mutase enzymes
35)Expanding functional protein sequence spaces using generative adversarial networks
36)Protein sequence design by explicit energy landscapeoptimization
37)De novo protein design by deep networkhallucination
38)Design of proteins presenting discontinuous functional sites using deep learning
39)Deep learning methods for designing proteins scaffolding functional sites
40)Low-N protein engineering with data-efficient deeplearning
41)Language models enable zero-shot prediction of the effects of mutations on proteinfunction
42)酶工程:从人工设计到人工智能
43)Protein sequence design with deep generativemodels
44)FLIP: Benchmark tasks in fitness landscape inference forproteins
45)MaveDB v2: a curated community database with over three million variant effectsfrom multiplexed functionalassays
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢