
【论文标题】De novo mass spectrometry peptide sequencing with a transformer model
【作者团队】Melih Yilmaz, William E. Fondrie, Wout Bittremieux, Sewoong Oh, William Stafford Noble
【发表时间】2022/02/09
【机 构】华盛顿大学、UCSD
【论文链接】https://doi.org/10.1101/2022.02.07.479481
【代码链接】https://github.com/Noble-Lab/casanovo
串联质谱法是分析复杂生物样品中蛋白质含量的唯一高通量方法,因此是推动蛋白质组学领域发展的主要技术。这个领域的一个关键挑战为识别负责产生每个观察到的光谱的多肽序列,而不利用多肽序列数据库形式的预先知识。尽管已经开发了各种机器学习方法来解决这个新的测序问题,但在对串联质谱进行建模时出现的挑战导致了结合多个神经网络和后处理步骤的复杂模型,这中间可能涉及到CNN、RNN、点云、动态规划、数据库搜索、预训练等等。本文提出了一种简单而强大的新肽测序方法Casanovo,它使用一个Transformer框架,直接从观察到的质谱峰的序列映射到氨基酸的序列。实验表明,Casanovo在使用标准的跨物种评估框架的基准数据集上实现了最先进的性能,并且也通过了分布外的样本,即具有从未见过的多肽标签的光谱进行的测试。Casanovo不仅实现了卓越的性能,而且只需要与其他方法相比更小的模型复杂度和推理时间。
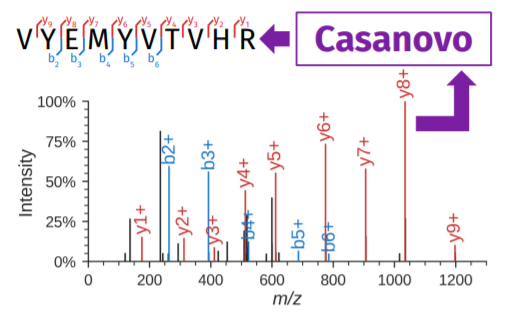
Casanovo执行从头开始的多肽测序。Casanovo将观察到的光谱作为输入谱并产生生成多肽的序列(VYEMYVTVHR)。在光谱中。与相关肽的b离子和y离子相对应的峰是彩色的,而黑色的峰则对应于意外碎裂事件或噪音。
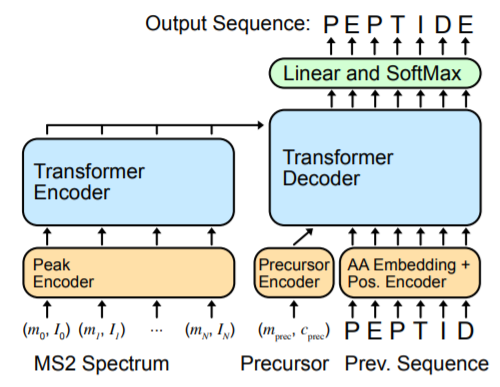
Casanovo的模型架构如上,其由Transformer编码器和解码器堆栈组成,它们分别负责学习输入频谱峰的隐层表征和解码频谱生成肽的氨基酸序列。谱系识别问题中,我们得到一个观察到的质谱和负责产生该谱系的肽(称为前体)的相关质量和电荷,需要推断出前体肽的氨基酸序列。编码器将d维频谱峰嵌入作为输入,并为每个峰输出d维隐层表征向量。随后,解码器将前面的氨基酸的表征作为输入,加上封装前体m/z和电荷信息的d维前体嵌入,来预测肽序列中的下一个氨基酸。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢