
2022年2月3日,西南交通大学计算机与人工智能学院的林小惠/江永全*/杨燕等人在Journal of Molecular Structure杂志发表文章,提出了一种基于图卷积网络预测原子间两两距离的模型,以解决传统计算方法在确定分子结构时实验成本高、计算成本高的问题。

论文链接:
https://authors.elsevier.com/a/1ebDZ54JURCNP
代码:
https://github.com/Lin12138xh/DMGCN
摘要
分子结构在许多领域有着重要的应用。例如,一些研究表明,在预测分子性质时,可以利用分子空间信息获得更好的预测结果。然而,传统的分子几何计算,如密度泛函理论(DFT),是十分耗时的。基于此,我们提出了一种基于图卷积网络预测原子间两两距离的模型,也称为分子距离矩阵预测(DMGCN)。为了说明DMGCN模型的效果,将该模型与DeeperGCN-DAGNN模型以及RDKit中计算分子构象的方法进行比较。结果表明,DMGCN的MAE小于DeeperGCN-DAGNN和RDKit。此外,将DMGCN模型预测的距离与QM9数据集计算的距离用于预测分子性质,从而显示了DMGCN模型预测的距离的有效性。
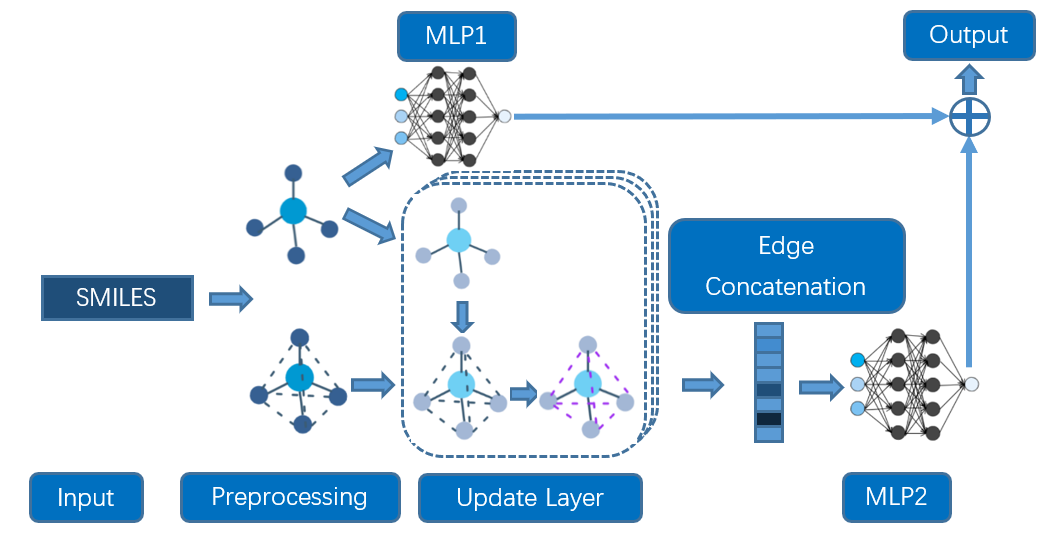
图1 模型架构。(1)预处理:根据输入的SMILES构建分子图和完全图;(2)更新层:通过分子图上的图卷积更新节点,并根据更新的节点更新完整图中的边。(3)边拼接:将每一更新层的边特征向量与上一更新层的节点特征向量进行拼接;(4)MLP1以拼接边特征向量和分子图中对应节点特征向量为输入,MPL2以拼接边结果为输入。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢