
解读作者:吴炜坤
深度学习一直被视为“黑箱操作”,而今天Sergey团队带来了其对Alphafold2结构预测机理的最新解读。
当代蛋白质结构预测的各类模型中,MSA信息提供了非常重要信息基础,直觉地,如果两个氨基酸残基在三维结构中存在稳定的相互作用,那在自然演化过程中,这两个位点的氨基酸类型的变化就会展现出协同变化的特性,从MSA中,已经发展出了各种方法来提取共进化信息帮助结构预测,包括alphafold2。
极度依赖MSA提供信息的预测模型并不利于预测孤儿序列,最近的一些方法,比如RGN2,试图通过利用深层语言模型对数百万个未标记的蛋白质序列进行训练来弥补这一缺陷,但这些模型在大多数蛋白质上的表现仍然明显低于AlphaFold,并且严重依赖MSA统计的信号。而在现实世界中,蛋白质的折叠仅依赖于其自身的序列,自发地快速地折叠成较低能量的构象状态,而不需要MSA的帮助。因此理想的蛋白结构预测模型应该就能从单序列中提取到足够的信息。
在传统蛋白折叠的方法中,研究者开发出了各类基于物理和统计理解的势能函数,结合构象搜索技术和能量评估、采样的技术来尝试解决蛋白质结构预测的问题,但这其中有2个明显的短板:不够准确的能量函数以及恐怖的蛋白质构象空间(Levinthal’s Paradox)。
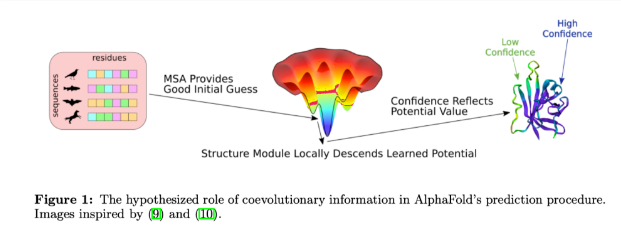
这不仅让人提出疑问,既然蛋白质的构象空间如此之大,为何Alphafold2能够单纯依赖共进化信息,就得出许多高精度的结构模型呢?

论文地址:https://www.biorxiv.org/content/10.1101/2022.03.11.484043v1.full.pdf
本文提出了一个alphafold2运行机制的大胆猜想:结构预测分为2个主要的步骤:
-
MSA的信息为模型提供了一个良好但模糊的猜测构象,从而在构象空间中定位到某些局部的极小值区间内;
-
AF2已经学习到了足够准确的能量函数,可以对猜测构象的质量好坏进行打分*(confidence score),从而经过多次迭代优化后得到可靠的结构。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢