
解读作者:吴炜坤
一、介绍
AlphaFold2(AF2)能够用于多肽-蛋白的PPI预测已经不是什么新鲜的事情了,通常的做法是添加长链甘氨酸或gap来欺骗模型,从而来做多肽复合物的“对接”预测。但美中不足的一点,在之前的实验中表明af2的plddt指标并不能准确反应多肽对靶点的结合力关系,这对AI多肽设计来说,是一个硬伤。
今天介绍的这篇文献做了一个非常有趣的实验,这个课题组之前是做增强采样的动力学模拟来研究多肽之间竞争/结合能力的评估,详见参考文献[1]。延续之前的研究,该团队尝试使用AlphaFold2来验证不同多肽对同个靶点蛋白的结合力趋势。具体的方法也很有意思,将体外模拟的竞争过程“代入”到深度学习模型中,简单来说就是同时地使用多个gap输入多条多肽和受体序列,让AF2预测这个混合物体系的具体构象!
二、计算结果
2.1 测试体系选择
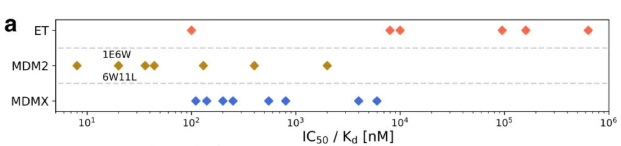
作者在之前的研究基础上,选择了ET domain和MDM2/MDMX-p53两个靶点作为研究对象,因为这个体系有跨度足够大的不同多肽序列以及晶体的结构。
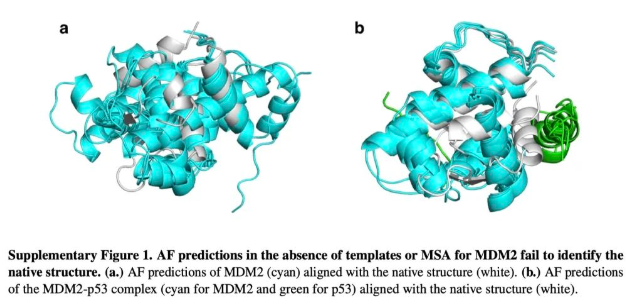
如果要准确地反映竞争模拟的过程,那前提条件是AF2能准确预测出这些多肽和受体结合的复合物结构。作者第一步测试了这3个靶点案例的预测情况。结果表明,对于MDM2/MDMX-p53体系,只需要使用受体MSA序列就够了。而ET体系有一条多肽预测失败(JMJD6),更进一步不提供MSA信息,AF2也成功预测了ET-peptide结合构象,但对于MDM2/MDMX-p53预测失败。
2.2 竞争模拟的方法
通过使用ColabFold预测受体-peptide1-peptide2的复合物结构,其中受体序列使用MSA信息(或附带模板结构信息),而多肽使用单序列信息,每个体系共计预测20次重复(5 models)。最后统计各种pose下多肽几何坐标中心与晶体的多肽构象的几何坐标中心的距离(也可以理解为简化版的RMSD)。在A100的加持下,每个体系的计算时间大约是4分钟。
2.3 竞争模拟效果
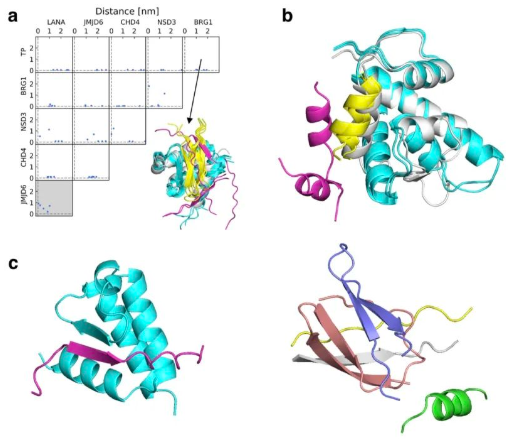
对于ET domain体系,作者采取两两组合的方式预测6个复合物体系的排名情况。AF2准确地预测出了6条多肽中3条的亲和力最强排名顺序(Kd: 0.1, 8 and 10 µM),而对于后三条结合力较差的多肽顺序不够明朗((Kd: 95, 160, 635µM)。通过构象分析,CHD4和LANA评估结合力类似,其主要构象为beta折叠片,并且受体之间的疏水相互作用要少得多。

在下图中可见TP这个结合最强的多肽在两两竞争的预测中,大多数的构象和真实构象的差异很小(distance = ~0 nm)。在BRG1中同样观察到了这个现象,说白了好的竞争体现在距离矩阵是否为接近0的直线。
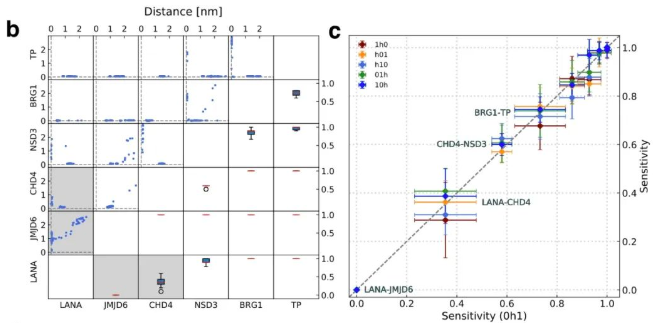
贴出3D结果图: 直观的意思是结合中心被强结合的多肽占据,而结合弱多肽的游离在外部。
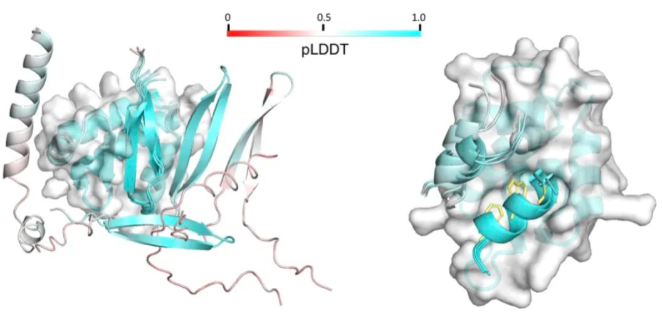
并且更重要的是,作者发现多肽的序列顺序对预测结果并没有影响。(0h1 stands for the input as peptide 0 :ET:peptide 1 .)
而在MDM2/X的体系中,多肽序列的相似性其实非常的高,也就是说关键的PPI残基并没有太大的变化。因此作者并不确定这种竞争的方法能否反映结合力,并且更糟糕的是,测试组中存在3条无活性的序列,AF2都错误预测了实际的复合物结构。因此AF2对突变不敏感的问题可能会在该方法中放大!
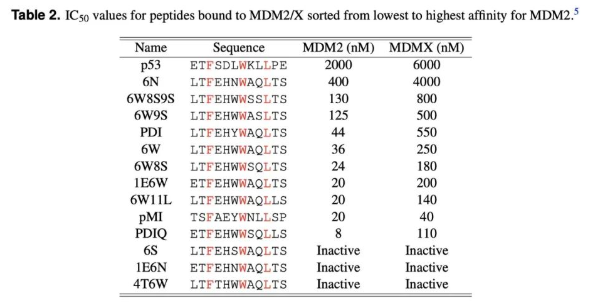
但令人兴奋的是,AF2虽然对突变不够完美,但能够正确地对几组多肽的结合力进行排名:同样PDIQ、6W11L、1E6W、PDI结合最强的多肽被筛选出来,而pMI、6W8S排名效果较差(每组测试中都存在2个cluster)。目前来看至少对亲和力存在几倍差异的案例有几率被识别出来。
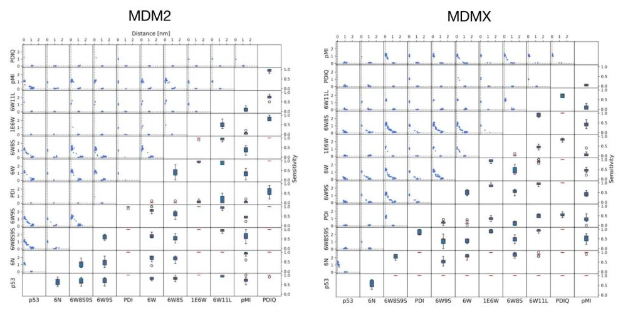
更进一步,作者尝试用这套方法查看能否正确排名多肽对MDM2和MDMX的选择性结合的排名。因为在实验中,这些多肽更倾向于结合MDM2。因此作者输入两条受体序列,分别评估每条多肽会竞争结合在哪个受体上。有趣的是,当提供了MSA信息,AF2预测的结构倾向于把两个受体dock在一起,因此作者选取了使用模板的方法,消除MSA的影响。结果表明11条多肽中,10条多肽都更加倾向于结合在MDM2,这个行为和实验的趋势一致。*(预印版本没有标准受体轴信息)
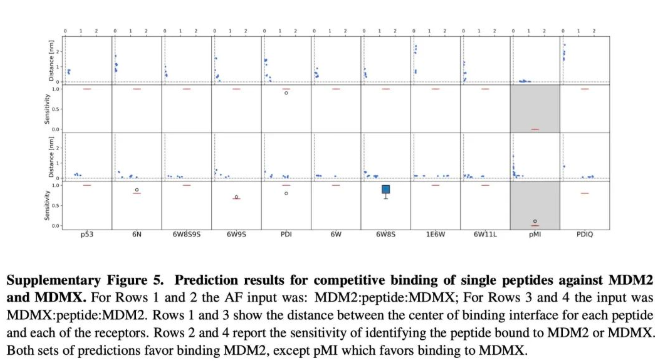
最后作者还做了多个多肽的竞争实验,结果表明多肽添加越多,预测的效率并不会得到大幅提升。但是可以依然可以确定出结合力最强的那个多肽的结合构象。对于ET domain的这个case,最强的多肽结合在活性中心,而其他的多肽形成组装体。
三、阿坤点评
作者通过拼接多段多肽来实现结合竞争的模拟思路较为新颖,并且也成功地识别出了结合最强的多肽序列。此意义对于线性螺旋肽的设计意义非常重大。因为在实际的药物设计过程中,我们只关心那些结合性质最好序列!并且AF2的预测时间较短,完全可以配合Rosetta等进行大量的多肽序列生成和设计。
但该方法应用也不是万能的,作者明确指出:
-
每条多肽体系必须单独被系统准确地预测出构象
-
多肽需要有结构化的二级结构(说白就是螺旋肽)
参考文献:
-
Morrone, J. A., Perez, A., MacCallum, J. & Dill, K. A. Computed Binding of Peptides to Proteins with MELD-Accelerated Molecular Dynamics. J. Chem. Theory Comput. 13, 870–876 (2017).
-
AlphaFold encodes the principles to identify high affinity peptide binders, Liwei Chang 1,2 and Alberto Perez1,2,* 【本文文献】
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢