

先导化合物的性质优化向来是药物研发过程中一个老大难的问题,人们既希望获得出色的靶点亲和力,又希望成药性相关的选择性、溶解性、毒性等各方面性质无一短板,每一项都不是简单的任务,联合起来就变得更为复杂。因此,真实的先导分子优化历程往往是“顾此失彼”,即使耗时耗力还不一定能获得理想的分子结构。面对这一行业难题,来自美国IBM研究中心的Payel Das团队提出了一种分子优化的通用型机器学习框架(QMO),像机械的关键通用零件一般可以与其他算法工具相互兼容,优化性能极佳,相关工作发表在Nature Machine Intelligence期刊上。

论文标题:
Optimizing molecules using efficient queries from property evaluations
论文链接:
https://www.nature.com/articles/s42256-021-00422-y
设计思路方面,QMO沿用了热门的机器学习思想,优点是可以从领域知识和生物大数据两方面获取有效信息,特别适合解决复杂的问题。具体的架构如图-1所示,QMO首先需要一个自编码器来编译分子,因为真实世界中的化合物结构空间过于庞大,通常被认为有多达10^60种可能,这里的自编码器可以将其压缩成低维且连续的空间,方便迭代过程中的结构采样工作。然后,研究者为自编码器的结构输出连接了自由的性质预测模块,用户可以按需接入各种成熟的算法工具,例如常用的有亲和力预测,成药性预测,和结构相似性预测等。而预测的结果会为QMO提供反馈,需要指出的是,这里一大特色是选取了零阶优化算法将预测反馈与自编码器组成框架的迭代闭环,不同于以往基于微分的优化思路,基于差分的零阶优化算法更加适合于处理非连续的数据类型,像化学分子的SMILES表示或者多肽的序列表示就是这样的类型。
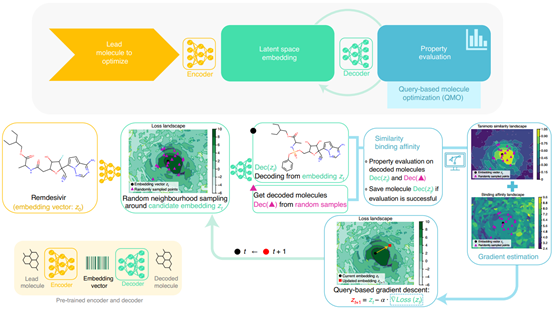
QMO框架的架构示意
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢