
【论文标题】Generative De Novo Protein Design with Global Context
【作者团队】Cheng Tan, Zhangyang Gao, Jun Xia, Stan Z. Li
【发表时间】2022/04/21
【机 构】西湖大学
【论文链接】https://arxiv.org/pdf/2204.10673v1.pdf
氨基酸的线性序列决定了蛋白质的结构和功能。蛋白质设计,被称为蛋白质结构预测的逆向,旨在获得一个新的蛋白质序列,使其折叠成定义的结构。最近关于计算蛋白质设计的工作研究了用局部位置信息为所需的骨架结构设计序列,并取得了有竞争力的性能。然而,不同骨架结构中类似的局部环境可能导致不同的氨基酸,这表明蛋白质结构的全局环境很重要。因此,本文提出了由局部和全局模块组成的全局背景感知生成式De nono蛋白质设计方法(GCA)。局部模块侧重于相邻氨基酸之间的关系,而全局模块则明确地捕捉非局部背景。实验结果表明,所提出的GCA方法在从头开始的蛋白质设计方面优于先进的方法。
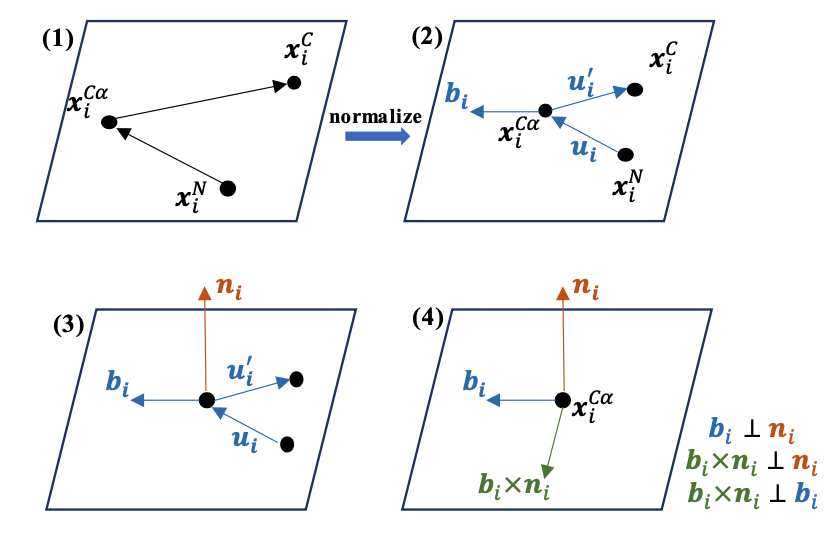
本文的蛋白质图构建中,node特征为残基3个二面角的sin与cos,edge特征只考虑C_alpha,包含三部分,1.残基间欧式距离的径向基函数以适应高斯分布;2.如上图所示的以i为中心的局部坐标系统转换,将残基间的单位向量转换到以[bi,ni,bi*ni]的局部三维坐标系中;3.以残基间坐标系之间旋转矩阵的四元数表征所代表的朝向信息。
该特征设置符合旋转平移不变性。其中节点特征只依赖于一个单一的节点,与其他节点没有任何联系,边特征也可证。
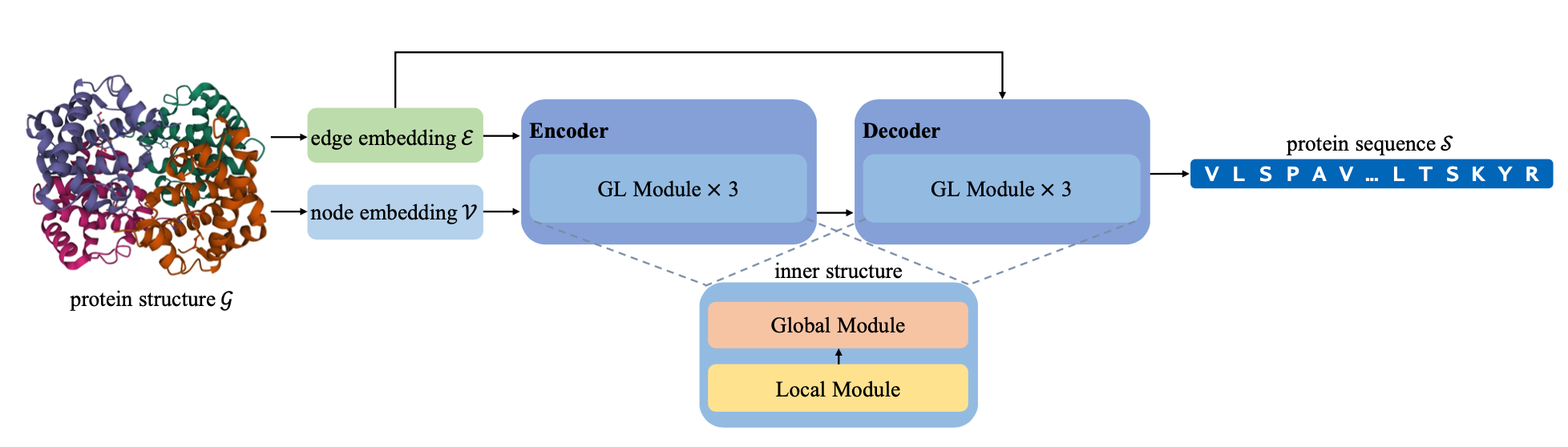
上图展示了GCA的框架,具有三个GL模块的编码器在edge嵌入的帮助下学习隐藏的node嵌入,具有相同的三块GL模块的解码器在结构和先前解码的氨基酸的条件下自动生成氨基酸。
模型带有注意力的GNN组成,其中的节点嵌入和边嵌入如上文所述,包括局部模块,其中注意力的范围由节点的KNN近邻组成,全局模块的使用了完整的自注意力网络,注意力范围为全部的节点,query信息为残基i节点信息,key信息包括残基i节点信息、残基j节点信息、残基i和残基j的边信息。
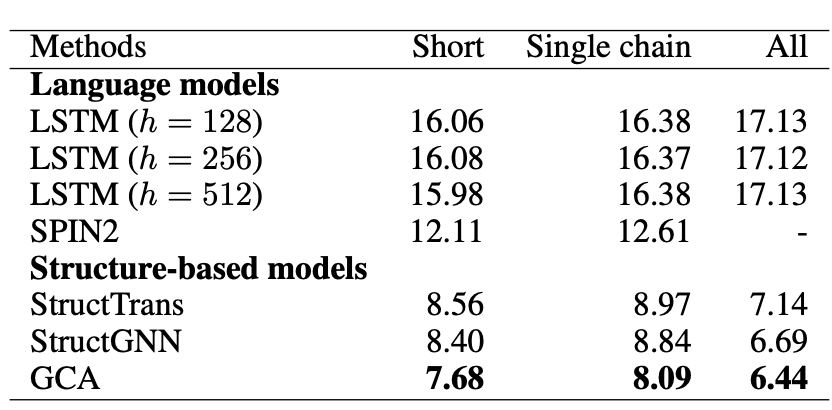
数据方面,本文包括三部分,1.使用CATH 4.2数据集来评估我们的方法在不同的蛋白质fold中的泛化能力,该数据集包括长度不超过500的多链蛋白,并且结构已经通过其CATH(类、结构、拓扑、同源)对所有域进行了40%的非冗余分类。测试集有两个子集,短序列子集,包括长度不超过100的链;单链子集,用于与只使用单链的基线进行比较。2.从AlphaFold DB中收集人类的蛋白质数据集。3.较小的数据集TS50,该模型仍然在CATH 4.2数据集上进行训练,我们对训练集和验证集进行过滤,以确保与TS50没有重叠。
评价方面,本文使用经典的perplexity和序列回复率,可以发现GCA在各个数据集上均领先。后续本文会在湿实验上证明模型。
创新点
- 本文分析了目前的计算蛋白质设计方法,一般方法忽略了全局语境。
- 本文提出了一种全局语境感知的方法,用于生成新的蛋白质设计,满足旋转和平移不变性。
- 在三个公共数据集上进行的广泛实验表明,GCA方法性能优越。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢