
【论文标题】Transferring Chemical and Energetic Knowledge Between Molecular Systems with Machine Learning
【作者团队】Sajjad Heydari, Stefano Raniolo, Lorenzo Livi, Vittorio Limongelli
【发表时间】2022/05/06
【机 构】曼尼托巴大学,卢加诺大学、埃克塞特大学、那不勒斯腓特烈二世大学
【论文链接】https://arxiv.org/pdf/2205.03339v1.pdf
预测分子体系的结构和能量特性是分子模拟的基本任务之一,它在化学、生物学和医学中都有应用案例。在过去的十年中,机器学习算法的出现影响了分子模拟的各种任务,包括原子体系的属性预测。本文提出了一种新的方法,将从简单的分子体系中获得的知识转移到一个更复杂的体系中,该体系拥有大量的原子和自由度。特别是,本文专注于高和低自由能状态的分类。本文的方法依赖于利用分子的新型超图(HNN)表征,编码所有相关信息以描述构象的势能,以及新型消息传递和集合层,以处理和预测这种超图结构的数据。尽管问题很复杂,本文的结果显示,从tri alanine到deco alanine体系的迁移学习的AUC为0.92。此外,本文还表明,同样的迁移学习方法可以在无监督的情况下用于将deco alanine的各种二级结构归入具有类似自由能值的群组。本文的研究可被视为一个概念证明POC,可以为分子体系设计可靠的迁移学习模型,为预测生物相关体系的结构和能量特性铺平道路。
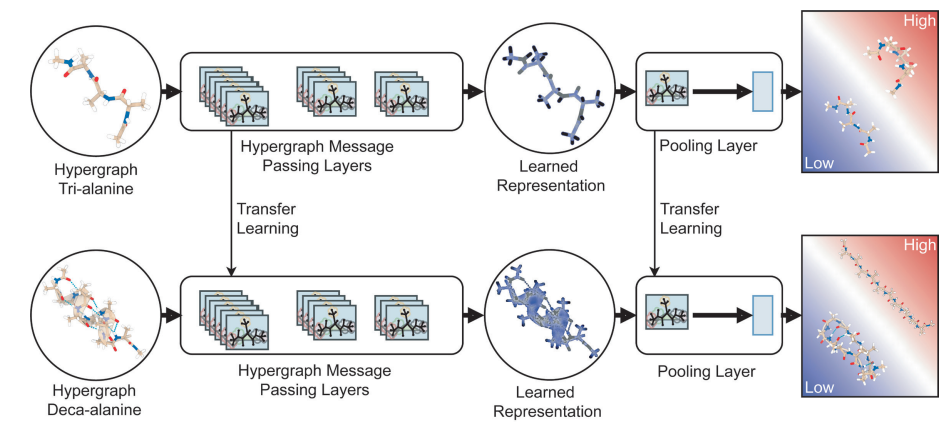
上图展示了迁移学习流程。图的上半部分代表了神经网络模型的训练,其中用于训练的分子的超图表示(如tri-alanine系统的例子)通过超图消息传递层获得隐藏表示。这种表征由一个集合层进一步处理,以输出输入为低能量状态的概率。图的下半部分描述了转移学习的过程,在这个过程中,训练好的模型被用来处理目标系统的例子(例如,deca-alanine)并作出相应的预测。
迁移学习是一种机器学习技术,用于在某些数据分布上学习模型,并在不同的分布上转移这些模型。它通常通过其源和目标分布以及源和目标任务来描述。目标是训练一个模型来解决源分布上的源任务,然后调整它,使其能够解决目标分布上的目标任务。在本文的实验中,本文考虑在源和目标分子系统之间的零样本迁移学习,零样本迁移学习并不假定在训练期间可以得到关于目标系统的任何信息,这使得它在本文感兴趣的分子动力学模拟环境中更有意义。本文感兴趣的任务是分类,特别是对在相关系统的自由能谱上对低能和高能状态进行分类。
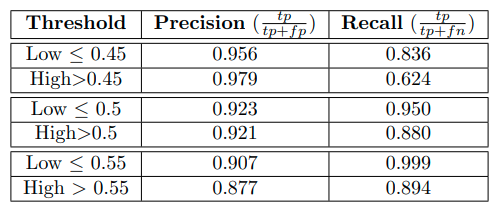
上图展示了deco alanine体系下不同阈值的分类结果,该模型在deco alanine系统上的表现取决于所选择的区分低能量和高能量状态的阈值。与前面的实验一样,为了提供一个不依赖于特定阈值选择的更稳健的分类性能指标,本文也进行了ROC分析并计算了模型的AUC,结果AUC高达0.92,从而证实了在考虑更复杂的迁移学习任务时,如在deco alanine系统中,也有可能以非常高的概率区分高能和低能状态。
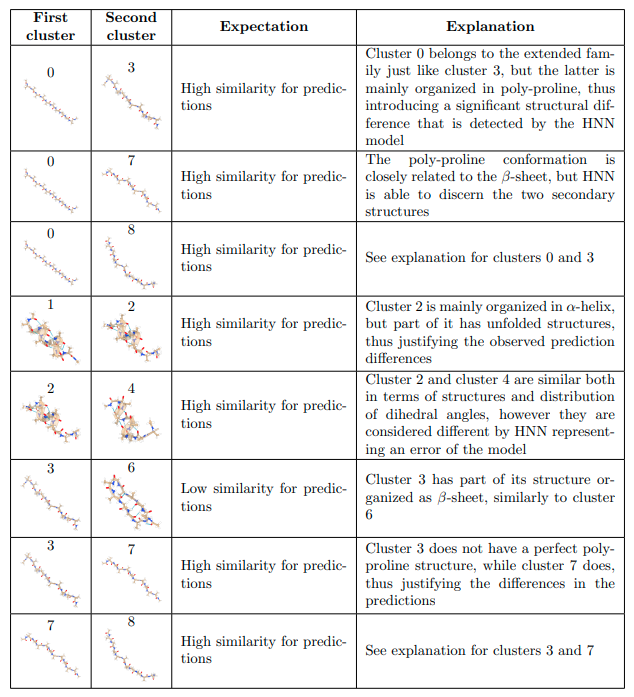
对HNN模型发现的结果的系统分析。Expectation一栏包含了基于聚类视觉比较的预期结果,而Explaination一栏则给出了偏离预期结果的理由,强调了模型如何能够检测出不同聚类中结构之间的微妙差异和相似性。
有趣的是,HNN模型能够识别属于同一家族的不同簇的结构,尽管有一些例外。例如,代表完美折叠的α螺旋结构的第1组被正确地识别为低能量状态,与第4组和第9组的结构相似,但与第2组不同。后者是这个家族的一个异类,从它的整体低p值可以看出,表明这样的一个簇与其他所有的簇有明显的不同。事实上,对第2簇和第4簇的比较表明,如果与第2簇相比,第2簇的C端最后三个残基处于相当不折叠的构象中,这两个簇的结构最接近。此外,根据通常使用的基于RMSD的结构标准,HNN能够检测到那些被认为非常相似的簇之间的细微差别。值得一提的是0号簇与发夹状家族的簇,即5号和6号簇的结果。对于发夹状家族的结构,两个β-片以反平行的方式组织,使分子内h-键的数量最大化。由于群组0是一个完全扩展的β结构(没有链间稳定的相互作用),HNN能够检测到类似特征的存在,但仍然能够区分群组。
创新点
迁移学习提供了一个框架,可以从大数据出发,对数据稀缺的问题进行预测,这样的框架已经被广泛使用。然而,到目前为止,它在计算化学中的应用很少,主要是用于近似量子力学计算或推断材料和分子特性。另一方面,据本文所知,通过机器学习评估构象取样中的自由能值是一个新颖而有趣的研究领域,科学界对它的兴趣越来越大.
在本文中,分子的结构特征与自由能估计配对,以便在转移学习中区分目标系统的高能或低能状态。这可能有很大的用途,因为它将完全取代冗长而昂贵的模拟,被机器学习模型所取代,一旦训练好,可以在一小部分时间内输出估计值。为了充分模拟分子系统的势能,从而达到预测其自由能的目的,需要考虑到两个、三个和四个原子之间的相互作用。
本文提出了一种新的基于超图的表示方法,使本文能够对所分析的分子系统的所有相关化学物理信息进行编码。在这方面,本文的工作代表了第一个使用超图来表示一个给定分子的所有必要的化学物理特性,从而标志着在机器学习和分子模拟领域的一个重大进步。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢