
【论文标题】RITA: a Study on Scaling Up Generative Protein Sequence Models
【作者团队】Daniel Hesslow, Niccoló Zanichelli, Pascal Notin, Iacopo Poli, Debora Marks
【发表时间】2022/05/11
【机 构】牛津、哈佛、lighton.ai
【论文链接】https://arxiv.org/pdf/2205.05789.pdf
【代码链接】https://github.com/lightonai/RITA
本文介绍了RITA:一套用于蛋白质序列的自回归生成预训练模型,有多达12亿个参数,在属于UniRef-100数据库的超过2.8亿个蛋白质序列上进行训练。这种生成模型有望大大加快蛋白质的设计。本文首次系统地研究了蛋白质领域的自回归Transformer的能力是如何随模型大小而发展的:本文评估了RITA模型在下一个氨基酸预测、零样本适应性和酶功能预测中的作用,显示了扩大模型的好处。
架构:为了给科学界提供一个尽可能普适的模型,本文选择将模型训练成没有任何调节信息的纯解码器Transformer模型。本文对位置嵌入技术进行了小规模的消融研究,本文评估了旋转位置嵌入(RoPE)和AliBi,并选择使用RoPE,因为由此产生的语言建模损失较低。本文训练了四个不同的模型,以研究模型大小和下游能力之间的关系,并使用与GPT-3相同的模型超参数和命名方案。
数据:为了保留预训练数据中包含的所有信息,本文选择在训练前不进行任何聚类。本文专注于三个不同的预训练语料库:UniRef100、MGnify和Metaclust,每个语料库都为模型预训练提供了足够数量的token,而不必重复数据。然后本文在短时间内训练三个小模型,以估计每个数据集对其他数据集的可转移性。实验表明,利用UniRef-100,其次是Metaclust,本文会得到最好的结果,而利用MGnify的结果最差,另外本文注意到,使用几个数据集的组合可能是有益的。
训练:本文利用Megatron-Deepspeed框架来实现高训练吞吐量,并使用数据和管道并行的组合来训练模型。
所有的模型都是在总共1500亿个氨基酸上训练的,训练运行是在IDRIS的Jean Zay超级计算机上进行的。这些模型的训练总时间超过25000个Nvidia-V100 GPU小时。本文利用Adam优化器),所有实验的批次大小为512,上下文大小为1024。
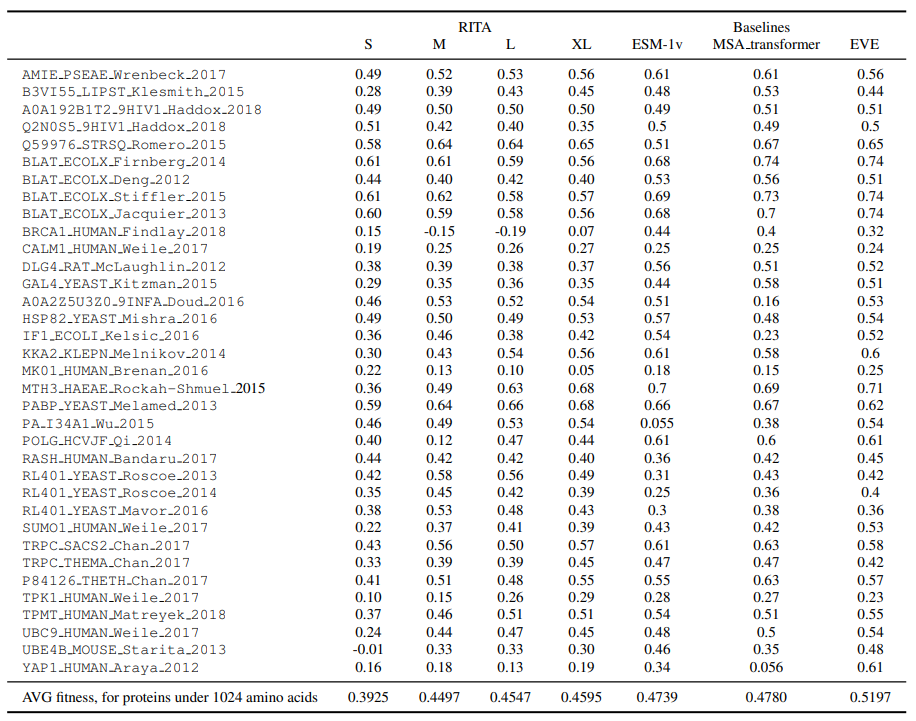
结果:上图展示了实验测量的不同蛋白质的fitness值与模型预测的值之间的Spearman相关性。本文将BRCA1 HUMAN、POLG HCVJF和UBE4B MOUSE从聚合中排除,因为它们不适合本文的模型所训练的上下文长度,而且旋转嵌入不能很好地推断。可以注意到,使用似然作为fitness代理是普遍的,不需要任何额外的信息,在这点上EVE模型有些不同。另外虽然ESM-1v和MSA Transformer在这个基准上表现较好,但在没有进一步监督的情况下,它们无法处理indels。
其他实验:语言模型可以通过在生成前加上人工创建的提示来解决各种各样的任务。这种做法得到了提示工程的名称,指的是寻找一个能产生令人满意的生成的提示的过程。prompt-funing,一种在嵌入空间中自动学习软提示的方法,已经成为执行参数高效微调的重要方式,只需学习微调通常所需参数数量的一小部分。本文研究是否有可能通过利用prompt-funing为预训练的蛋白质序列模型增加可控生成。本文任意选择了一个在训练中被保留下来的蛋白质家族,PF03272,并学习了一个专门生成这个家族的蛋白质的提示。最后看到在prompt-funing下,困惑度明显降低,表明该模型确实能够学习生成这个蛋白质家族的蛋白质。
创新点
- 本文介绍了RITA1,这是一个用于蛋白质设计的生成性蛋白质序列模型系列,参数多达1.2B。
- 本文研究了模型大小和下游任务性能之间的关系,为蛋白质序列建模的扩大规模迈出了第一步。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢