
【论文标题】Masked inverse folding with sequence transfer for protein representation learning
【作者团队】Kevin K. Yang, Niccolò Zanichelli, Hugh Yeh
【发表时间】2022/05/28
【机 构】微软、芝加哥大学、EleutherAI
【论文链接】https://doi.org/10.1101/2022.05.25.493516
【代码链接】 https://github.com/microsoft/protein-sequence-models
对蛋白质序列进行自监督的预训练,使蛋白质功能和适应度预测的性能达到了一流水平。然而,仅有序列的方法忽略了实验和预测的蛋白质结构中所包含的丰富信息。同时,反折叠方法根据蛋白质的结构重建了蛋白质的氨基酸序列,但没有利用没有已知结构的序列的优势。在这项研究中,作者训练了一个被遮蔽的反折叠蛋白质语言模型,其参数为一个结构图神经网络。本文表明,使用预训练的仅有序列的蛋白质遮蔽语言模型的输出作为逆折叠模型的输入,可以进一步提高预训练的复杂度。本文在下游的蛋白质工程任务中评估了这两个模型,并分析了使用来自实验或预测结构的信息对性能的影响。

上图展示了3种模型:蛋白质的卷积自编码遮蔽语言模型(之前已经发过介绍),遮蔽反折叠模型,和带有序列迁移的遮蔽反折叠模型。
遮蔽反折叠是一个预训练任务,旨在重建一个以其骨架结构为条件的被破坏的蛋白质序列,这里使用BERT破坏方案并训练模型以重建以破坏的序列和骨架结构为条件的原始氨基酸。经过预训练后,MIF模型可用于执行纯序列MLM所能执行的任何下游任务,但必须提供一个结构。
MIF-ST加入了序列迁移,虽然与仅有序列的预训练相比,对结构的条件提高了序列的回复率,但本文假设,从没有结构的序列中迁移信息应该进一步提高预训练任务的性能。因此,作者通过直接用CARP-640M在UniRef50上预训练的输出来取代序列嵌入来迁移序列信息。MIF-ST是用与MIF相同的超参数训练的。最后的结果展示在CATH4.2测试集上,序列迁移比CARP-640M和MIF都提高了困惑度和回复率。使用随机初始化权重的CARP-640M架构作为MIF的输入并没有改善性能,这表明仅仅增加模型容量是不够的,序列迁移是改善的必要条件。

上图展示了零样本预测,具体数据集描述如下:
- DeepSequence。41个深度突变扫描数据集,这些数据集分别测量了成千上万的突变或突变组合对母体序列的影响。对模型的评估是根据它们在数据集上的平均Spearman ρ。本文使用了每个野生型蛋白质使用ColabFold默认参数获得的五个AlphaFold结构的结果。
- 突变稳定性预测(MSP):SKEMP的单一突变体,衡量突变体蛋白质是否显示出比野生型更好的结合。在测试集的893个阳性例子和3255个阴性例子上,使用接收操作曲线下的面积(AUROC)对模型进行评估。
- 稳定性:使用深度突变扫描来测量一组从头设计的具有10种不同fold的小蛋白的单点突变体的蛋白酶稳定性。对模型的评价是根据它们在不同折叠中的平均皮尔逊相关度。
- RBD:深度突变扫描测量SARS-CoV-2尖峰蛋白的受体结合域(RBD)的所有氨基酸突变如何影响其对人类ACE2受体的亲和力。因此,模型的任务是预测所有1311个突变的RBD序列的结合亲和力。本文使用PDB 6M0J作为原始结构。
- GB1:对蛋白G域B1进行了4个位点的组合式深度突变扫描,这是一种在链球菌中表达的免疫球蛋白结合蛋白。使用56个氨基酸的GB1结构域的AlphaFold结构,模型进行了斯皮尔曼相关系数评估。
从结果上来看,对于除DeepSequence之外的所有任务,MIF都优于纯序列方法,而在DeepSequence上,增加序列迁移后的性能高于纯序列方法。在DeepSequence中,MIF-ST在41个数据集中的22个击败了CARP-640M,在41个数据集中的37个击败了MIF。在其他任务中,MIF和MIF-ST取得了相似的结果,尽管预训练性能有所提高,但序列迁移并没有持续改善零样本性能。MIF在稳定性数据集测试的所有10个折叠上都优于CARP-640M,而MIF-ST在10个折页中的6个折页上优于MIF。在MSP和稳定性方面,MIF和MIF-ST与其他反折叠方法相当,但GVP+AF2在RBD上明显胜出。
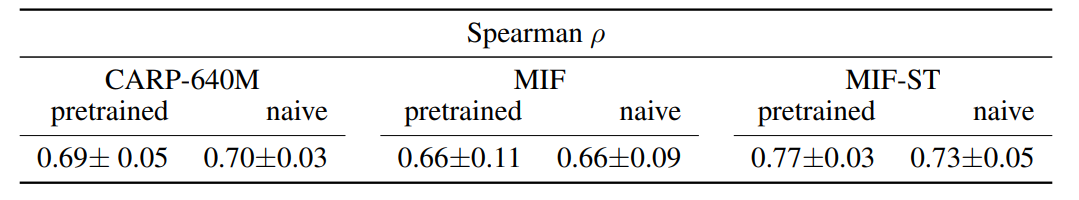
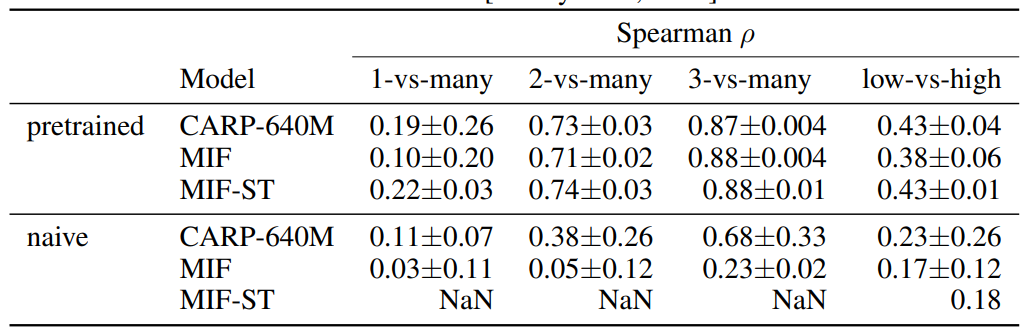
除了零样本预测外,预训练的蛋白质模型最好能够做出在蛋白质工程活动中经常出现的域外预测类型。本文在两种适应度景观上进行微调和评估。
- Rma NOD:Rhodothermus marinus(Rma)一氧化二氮二氧酶(NOD)酶的七个位置的突变如何影响苯基二甲基硅烷与2-重氮丙酸乙酯反应的对映选择性。本文对214个在3个位置发生突变的变体进行训练,并将在所有7个位置发生突变的变体随机分成40个验证变体和312个测试变体。这测试了该模型在一个小的训练集的基础上对未见过的位置上的突变的归纳能力。本文使用PDB 6WK3作为结构。测量结果从ProtaBank检索。
- GB1。本文使用FLIP]在GB1景观上的拆分。这些拆分测试了从较少突变到较多突变或从较低适应度序列到较高适应度序列的概括性。本文使用PDB 2GI9作为结构。
在Rma NOD任务上,MIF-ST架构上的表现优于CARP-640M和MIF,无论是否有预训练,MIF-ST也是唯一一个通过预训练提高该任务性能的模型。
在GB1任务上,所有模型都从GB1任务的预训练中受益。具有随机权重的MIF-ST一直收敛到一个退化的解决方案,即它对测试集中所有3个随机种子的所有序列在4次分割中的3次都预测了相同的值。将结构调节与序列迁移相结合似乎略有帮助,当训练集被限制为单一和双重突变体时,收益最大。有趣的是,如果没有预训练,增加结构信息并不能提高性能。
创新点
- 在这项工作中,本文对1.9万个结构和序列的遮蔽反折叠进行了研究,作为预训练任务。
- 本文观察到,MIF是一个有效的预训练模型,适用于各种下游的蛋白质工程任务。
- 本文通过迁移在数千万蛋白质序列上训练的模型的信息来扩展MIF,改善了预训练的困惑性和一些下游任务的性能。
- 来自AlphaFold的高质量的预测结构往往比实验结构的零散性能有所提高。
- 有趣的是,改善预训练的迷惑性并不总是能带来更好的下游性能。
展望
- 相关工作部分提出了应可与MIF和MIF-ST相结合的改进。使用更先进的GVP或SE(3)变换器架构,而不是结构化GNN作为基础模型,可能会改善预训练性能,就像使用AlphaFold结构或向输入结构添加噪声一样。
- 另一个明显的扩展是用自回归或Span遮蔽损失进行训练,这应该更适合于生成任务,更好地处理插入和删除,并可能更好地推广到复合物。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢