
【论文标题】Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval
【作者团队】Pascal Notin, Mafalda Dias, Jonathan Frazer, Javier Marchena-Hurtado, Aidan Gomez, Debora S. Marks, Yarin Gal
【发表时间】2022/05/27
【机 构】牛津、哈佛
【论文链接】https://arxiv.org/pdf/2205.13760v1.pdf
【代码链接】 https://github.com/OATML-Markslab/Tranception
【备注】ICML2022
准确模拟蛋白质序列的适应度景观的能力对于从量化人类突变对疾病可能性的影响,到预测病毒中的免疫逃逸突变和设计新型生物治疗蛋白的广泛的应用至关重。在多重序列比对上训练的蛋白质序列的深度生成模型是迄今为止解决这些任务的最成功方法。然而,这些方法的性能取决于是否有足够的深度和多样性的比对来进行可靠的训练。因此,它们的潜在应用范围受到了许多蛋白质家族很难比对的事实的限制。在来自不同家族的大量未比对的蛋白质序列上训练的大型语言模型可以解决这些问题,并显示出最终弥补性能差距的潜力。本文展示了Tranception,这是一种新型的Transformer结构,利用自回归预测和推理时同源序列的检索来实现最先进的适应度预测性能。鉴于其在多个突变体上的明显更高的性能,对浅层比对的鲁棒性和对缩略词的评分能力,本文的方法提供了比现有方法更重要的范围。为了在更广泛的蛋白质家族中进行更严格的模型测试,本文开发了ProteinGym--一套广泛的突变体效应的多重检测方法,与现有的基准相比,大大增加了检测方法的数量和多样性。
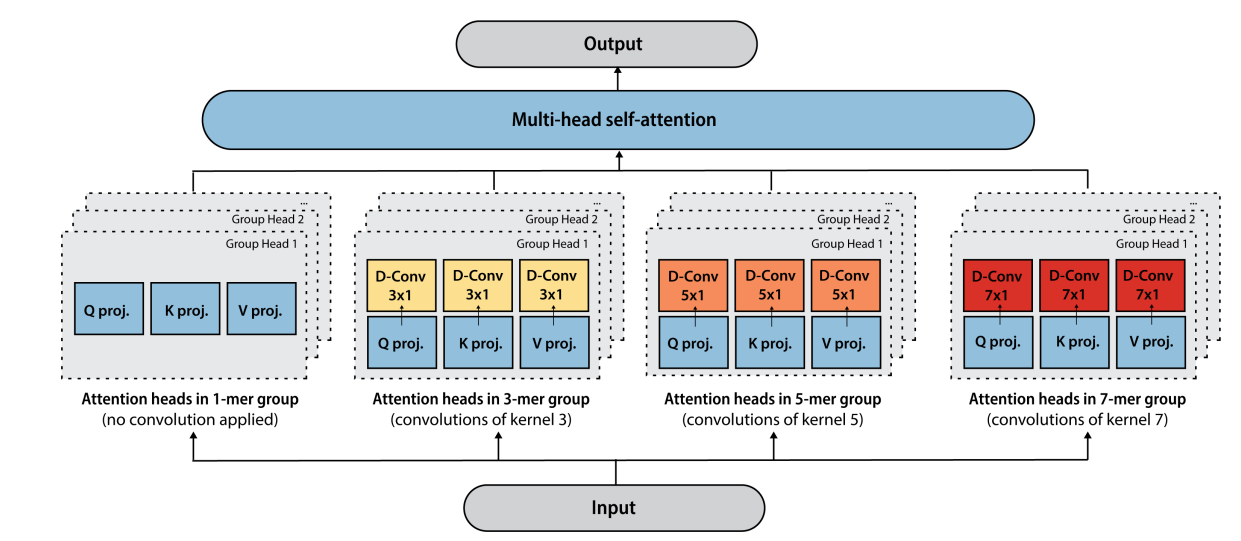
上图展示了Tranception注意力机制,该方法结合Primer和Inception网络的思想,研究明确关注氨基酸标记的连续子序列的好处。与Primer类似,本文在不同的多头注意力投射后利用平方ReLU激活和深度卷积。本文没有对每个深度卷积使用相似大小的核,而是将每层的注意力头分成4组,除了第一组不混合不同标记的信息(即 "1-mer "组),其他3组的Q、K和V投影后,分别应用独立的空间深度卷积,内核大小为3、5和7,从而像Inception一样将不同分辨率的信息结合起来。这激励了每个注意力头在整个序列长度的不同范围内混合信息,与Primer和GPT2相比,这导致了更有效的训练和下游任务表现。
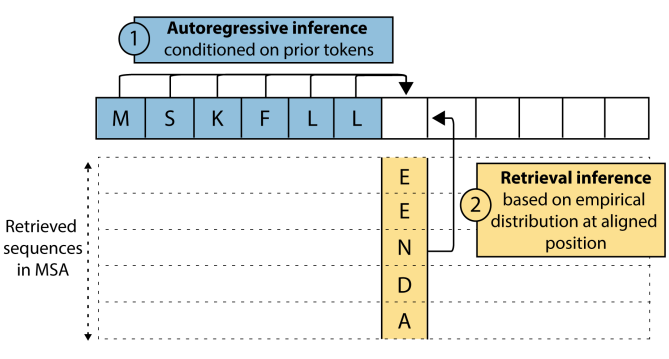
上图展示了结合自回归推理和检索推理的方法。Tranception中的预测是基于两种互补的推理模式:基于先前生成的标记的上下文的自回归预测和基于检索到的同源序列集合中每个位置的氨基酸的经验分布的预测。步骤如下:
第一步包括在推理时间为感兴趣的蛋白质家族的野生型序列检索MSA。当关注氨基酸替换时,检索到的同源序列集对野生型和突变序列都是通用的:本文对每个家族做一次检索步骤,并对所有要评分的突变序列摊销成本。当处理插入和删除时,本文通过删除MSA中与删除位置相对应的列,并在MSA中增加突变蛋白中插入位置的零填充列,使检索到的MSA适合每个突变序列。在推理时,插入的列或完全不覆盖的位置被忽略,模型只依靠其自回归模式在这些位置进行预测。
第二步是根据伪计数(忽略gap)和拉普拉斯平滑法计算每个比对位置的氨基酸经验分布。由于蛋白质数据库中发现的序列分布因人类取样和进化取样而有偏差,本文重新权衡MSA中的序列。最后,本文通过来自自回归推理模式的对数似然log PA(x)和来自检索推理模式的对数似然log PR(x)的加权算术平均值来估计一个蛋白质序列x的对数似然log P(x)。这可以等效地看作是概率空间中的加权几何平均,并形成一个适当的概率分布。
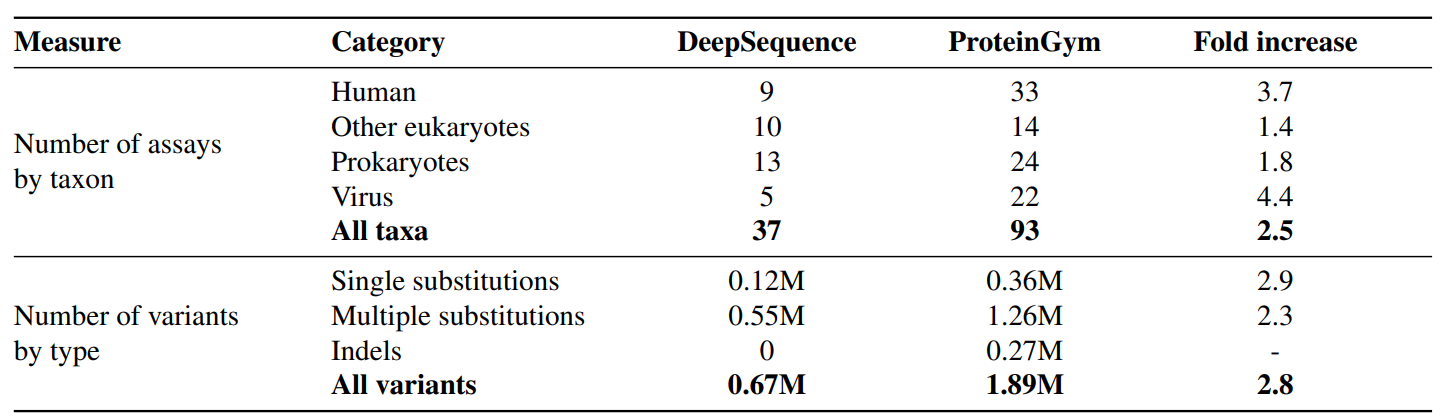
ProteinGym是一套广泛的最大和最多样化深度突变扫描(DMS)试验,旨在对不同制度下的各种突变效应预测器进行彻底的比较。它包含的实验和变体数量是DeepSequence基准的两倍多,该基准是为同一目的创建的。ProteinGym由两个基准组成:1)替换基准,包括87个DMS检测中的150万个错义变体的实验特征;2)indel基准,包括7个DMS检测中的30万个突变体。
实验测量的蛋白质适应性与进化选择的序列分布所反映的关系是复杂的。自然发生的蛋白质的适配性是这些蛋白质在生物体内受到的一套复杂的重叠约束的结果。因此,要确定一个既易于实验测量又能反映这种复杂性的单一分子特性,往往是具有挑战性的。因此,为了建立这个基准,本文优先考虑了一些实验,在这些实验中,每个突变的蛋白质的实验测量的特性都有望反映出该蛋白质在生物体内的作用,以及通过实验重复测量的质量。由此产生的DMS试验涵盖了广泛的功能特性(例如,热稳定性、配体结合、聚集、病毒复制、耐药性),跨越了不同的蛋白家族(例如,激酶、离子通道蛋白、G-蛋白偶联受体、聚合酶、转录因子、肿瘤抑制因子)和不同的类群(例如,人类、其他真核生物、原核生物、病毒)。
虽然大多数策划的DMS检测(在本文的基准或其他)探测单氨基酸替换的影响,但本文的集合还包括几个多氨基酸变体,这对于评估模型在序列空间中进一步推断它们所训练的自然发生的蛋白质的能力至关重要。最后,由于大多数突变效应预测器不能量化插入和缺失的影响,因此在大多数先前的基准中没有缩进。本文在Shin等人和Dallago等人提供的DMS集上进行扩展,以解决这一问题。蛋白质功能和生物体健身之间的关系已被证明往往是非线性的。因此,本文使用模型得分和实验测量之间的Spearman等级相关系数作为衡量模型性能的标准。在某些情况下,DMS的测量结果具有双峰的特点,等级相关并不适合。为此,本文提供了额外的模型性能测量:ROC曲线下的面积(AUC)和模型得分与实验测量之间的马修斯相关系数(MCC)。
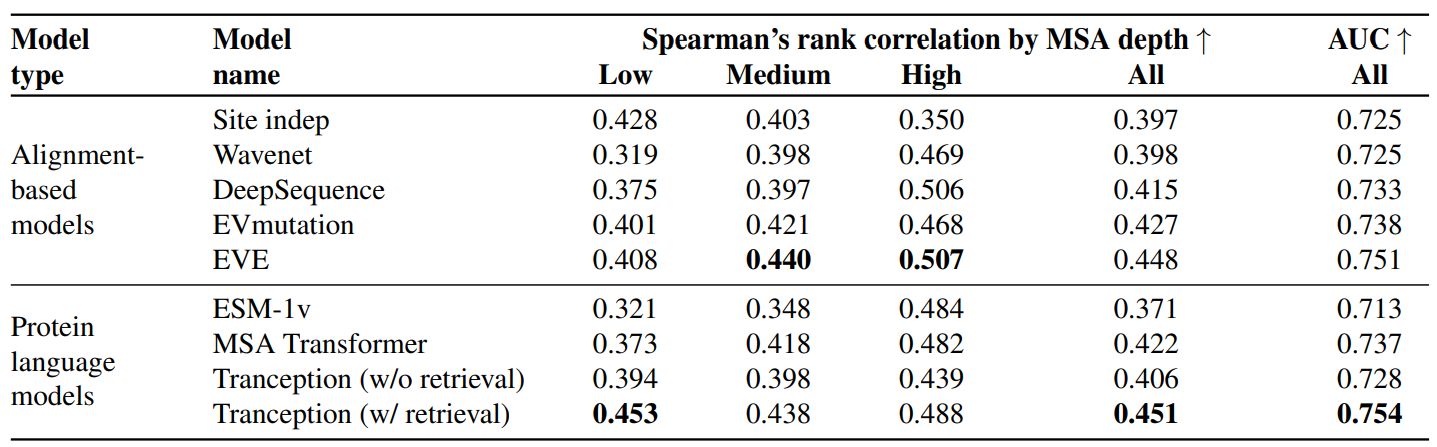
上图展示了在ProteinGym替换基准上,按MSA深度计算的模型得分和实验测量值之间的平均AUC和Spearman等级相关性。各种蛋白质的比对深度由MSA中的有效序列数Neff的比率来衡量,带检索的Tranception在整体基准上优于所有其他基线,在低深度MSA和多突变体的制度下,性能明显较高。在分析分类群水平的性能时,我们观察到Tranception在不同类别中的性能始终很高,特别是在人类蛋白质和其他真核生物上。这种在人类蛋白质上的高表现有着直接的临床应用,因为Tranception的性能优于EVE,并直接扩展到整个人类蛋白质组的建模,而EVE模型需要为每个感兴趣的新蛋白质进行训练,在撰写本文时只有3千种蛋白质可用。在没有检索的情况下,Tranception的性能优于ESM-1v。它是唯一的基线,也没有利用比对进行推理。在具有浅层比对的蛋白质上,在多重突变和病毒性蛋白质上,性能提升尤其明显。
另外本文报告了ProteinGym indel基准的性能指标(Spearman,AUC,MCC),并将Tranception的性能与Wavenet进行了比较,Wavenet是唯一能够量化删除或插入效果的基线。其他基于比对的模型受到它们所训练的原始MSA的固定坐标系统的限制。ESM-1v和MSA transformer都依赖于一种评分启发法,要求突变的位置存在于野生型序列中。在indel基准测试中,Tranception在有检索和无检索的情况下都优于Wavenet。
创新点
- 本文介绍了Tranception,这是一种新型的自回归Transformer结构,可以促进各注意头的专业化,以增强蛋白质建模。
- 本文在推理时将自回归预测和检索到的序列的同源性结合起来,以达到最先进的关于替换和插入删除的适应度预测性能。
- 本文整理了一套泛用的突变体效应的多重检测ProteinGym基准,与现有基准相比,大大增加了检测的数量和多样性。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢