
【论文标题】Towards generalizable prediction of antibody thermostability using machine learning on sequence and structure features
【作者团队】 Ameya Harmalkar, Roshan Rao, Jonas Honer, Wibke Deisting, Jonas Anlahr, Anja Hoenig, Julia Czwikla, Eva Sienz-Widmann, Doris Rau, Austin Rice, Timothy P. Riley, Danqing Li, Hannah B. Catterall, Christine E. Tinberg, Jeffrey J. Gray, Kathy Y. Wei
【发表时间】2022/06/07
【机 构】约翰霍普金斯、加州伯克利、安进公司
【论文链接】https://doi.org/10.1101/2022.06.03.494724
在过去的三十年里,单克隆抗体(mAb)作为治疗药物一直在备受关注,FDA最近批准的第100个mAb的里程碑式的结果就是证明。与结合单一目标的mAb不同,具有单链可变片段(scFv)模块的多特异性生物制剂(bsAbs)由于具有接触不同目标的优势而获得了特别的关注。尽管它们具有精致的特异性和亲和力,但这些scFv模块相对较差的热稳定性往往阻碍了它们作为潜在治疗药物的发展。近年来,通过突变来提高抗体序列的稳定性的工程已获得相当大的发展。由于抗体工程的实验方法费时、费力、费钱,计算方法可作为常规途径的快速和廉价的替代方法。这项工作展示了两种机器学习方法,一种是用预训练的语言模型(PTLM)捕捉序列变异的功能效应,第二种是用Rosetta能量特征训练的监督卷积神经网络(CNN),以更好地从序列中对热稳定scFv变体分类。这两个模型都是通过来自多个scFv序列库的温度特定数据(TS50测量值)来训练的。本文表明,一个用能量特征训练的简单CNN模型比预训练的语言模型在分布外序列上的泛化性更好(平均Spearman相关系数为0.4,而不是0.15)。此外,本文证明,对于一个独立的具有20个热稳定实验结果的的mAb,这些在TS50数据上训练的模型可以识别18个残基位置和5个相同的氨基酸突变,显示出显著的泛化能力。本文的结果表明,这种模型可以广泛适用于改善抗体的生物学特性。此外,将这类模型用于scFvs的其他物理化学特性,在优化mAbs或bsAbs的大规模生产和传递方面有潜在的应用。
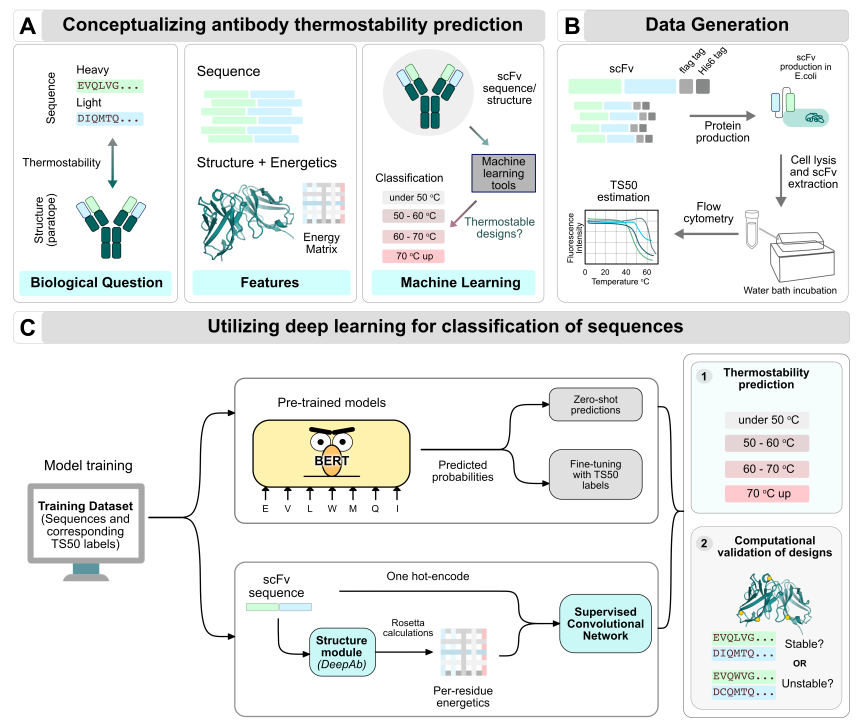
上图展示了使用深度学习识别scFv热稳定性的流程。
(A) 从序列中预测抗体的热稳定性。抗体的热稳定性与抗体在下游过程的可开发性息息相关。这一挑战的现有数据可以包括氨基酸序列、结构和计算出的能量。序列层面的热稳定性预测的生物学问题涉及识别哪些序列可以导致高度热稳定性的生物分子结构;对于抗体和scFvs,这意味着在高温压力下折叠结构状态和/或抗原结合的保存。蛋白质的热稳定性取决于残基级的生物物理属性。但是,对于抗体和scFvs来说,由于序列中的高度共识,破译仅基于序列模式的启发式或经验式规则来区分热稳定和不稳定的序列是具有挑战性的。机器学习(ML)模型已经显示出在没有潜在的生物物理途径的情况下提取映射序列与功能的高阶关系的潜力,并且它们在分类任务中表现良好。利用序列和结构信息作为特征,应用于大量预测任务的ML方法,如荧光性、内在稳定性、错义变体效应、蛋白质适应度、抗原特异性,已经显示出很高的成功率。
(B) 热稳定性数据的生成。本文从各种工程研究中收集温度数据,以开发热稳定性的scFv分子。这些序列数据包含通过对多个种系的重链和轻链进行突变而组装的scFv序列。来自17个种系的2700个scFv序列被整理构成序列数据。此外,来自另一项scFv研究的序列和一个孤立的scFv数据集构成了分布外数据集。对于每个序列,用TS50测量来评估热稳定性,代表目标结合的半最大值的温度。对于独立的scFv数据集,热稳定性是通过Tm测量来评估的,它代表了随着温度升高从折叠到展开的第一个转折。
(C) 训练一个预测TS50的分类网络。其中一种方法是用无监督的模型进行迁移学习。本文利用预训练的类似BERT的模型(如ESM1-b、ESM1-v等),用标记的TS50数据集进行零样本预测和微调预测。另一种方法是用监督卷积模型,利用注释的热稳定性数据,在序列水平(即单次编码的氨基酸类型)和能量水平(即用DeepAb(23)生成的假定三维结构模型获得的热力学能量)上配备简单的深度卷积网络,进行特征编码。通过这两个模型,本文的目的是预测一个给定scFv序列的热稳定性。通过开发一个可以有效过滤和筛选scFv序列的模型,本文可以大大加快识别更好的变体,以获得稳定的、可制造的、多特异性的生物制品。

上图展示了对预训练的无监督模型进行微调,提高了与目标的相关性。
A图、C图和E图展示了用预训练的模型进行基于零样本的似然预测,结果上与TS50数据集、分布外数据集的相关性不强。
B图、D图展示了在TS50数据上对预训练模型进行微调,明显改善了与目标的相关性。
F图展示了在TS50数据上微调的模型不能很好地推广到分布外数据集。

上图展示了能量特征可以提取热稳定性的 "通用 "信息。
A图展示了有监督的CNN用于对抗体序列进行分类的流程。这样的动机是,Shanehsazzadeh证明了小型监督模型可以在TAPE基准的下游预测任务中取得有竞争力的性能。类似的预测性能也被报道为使用监督卷积网络进行抗原特异性预测。
在该流程中,输入的scFv序列通过DeepAb的结构生成模块,然后是基于Rosetta的能量评估(总能量分为单体i-i和双体i-j残基能量,第i个残基与第j个残基的能量贡献制成表格),以估计scFv结构中每个氨基酸残基的每一残基能量。序列是one-hot编码的,能量特征以i-j矩阵表示,提供给网络。序列分支和能量分支的输出被串联起来,并通过密集层进行扁平化处理,输出序列在每个温度段的概率。
B图展示了由纯序列模型和纯能量模型学到的表征,通过t-SNE将每个序列的密集层的嵌入投射到两个维度。彩色点的聚集表明,仅有序列的模型嵌入按其实验集进行了聚类。另一方面,仅有能量的模型嵌入与实验集无关。因此,尽管有一个序列多样化的数据集,但仅对序列特征进行训练的微调和监督模型能够推断出序列的基本实验来源,并使热稳定性预测出现偏差,使其对较新的、分布外的数据集的泛化能力降低。
C图展示了70度以上分类的AUC曲线。由于仅有能量的模型相对来说更具有普遍性,本文通过构建一个由70-up的预测得出的ROC曲线来评估这个监督模型的性能。由于本文的目标是识别热稳定的序列,70-up的预测精度是最重要的。本文评估了四个测试数据集的ROC:两个保留数据集(P组和Q组)和两个代表一个测试scFv和一个独立scFv的分布外数据集。ROC下的面积超过0.7,表示分类精度很高。
D图显示了所有四个测试数据集的Spearman相关系数,分别与纯能量、纯序列和能量+序列模型的相关系数。在保留的数据集(P组和Q组)上,仅能量学模型的系数超过0.5,能量学+序列模型的性能同样较好。但是在分布外数据集上,能量学+序列和纯序列的性能下降(系数低于0.1)。纯能量学模型在盲数据集上仍显示出相对较高的相关性(分别为0.2和0.4)。

上图展示了对重链和轻链的实验性热稳定突变体的70-up预测与两种模型的对比,对一个抗体FV片段的计算DMS显示与实验性热变性数据一致。
用本文的监督网络验证所有的抗VEGF抗体的点突变体(PDB ID:2FJG(结合)和PDB ID:2FJF(非结合)),本文观察到在超过70ºC中预测的突变体与之前工作中的实验热变性数据有协同作用。球体表示经实验验证的提高Tm的突变体;粉色表示来自网络的预测,具有相同的残基位置,但不同的氨基酸突变;红色表示与实验数据相匹配的预测,灰色表示在计算预测中没有观察到的突变。缩略图突出了与实验和潜在相互作用一致的突变。表中说明了与实验和计算预测的比较。另外在分析的4540个点突变中(Nres=227个残基,每个残基20个氨基酸),只有20个点突变的实验数据可用。由于只有0.44%的抗血管内皮生长因子抗体可能的突变是通过实验来评估熔化温度的,所以热稳定性的验证数据集是非常少的。此外,尽管是温度特定的属性,TS50和Tm是不同的实验测量,并不完全相关。
通过实验验证的20个突变位置在卡通图中以球体形式突出显示。本文的网络正确识别了20个突变中的5个。令人惊讶的是,这五个突变都是由框架区残基组成的。此外,对于20个突变中的18个,SCNNs可以正确识别残基位置,尽管预测不同的氨基酸突变是最热稳定的。本文的网络可以在90%的情况下预测热稳定性的残基位置,其中25%是成功的预测(正确的残基位置和氨基酸残基)。通过将在TS50测量中训练的网络外推到其他的热聚集实验中(在这种情况下是Tm),本文证明内在的热属性可以被这种模型所捕获。
创新点
首先本文通过预训练的语言模型来装备无监督的学习表征,将序列分类到温度特定的类,量化其热稳定性。与使用序列或结构坐标特征的传统机器学习方法不同,本文将丰富的信息与热力学特征相结合。
此外,本文在小型、有监督的CNN模型上测试了使用能量特征进行分类任务的性能。
最后,本文通过识别实验验证的抗血管内皮生长因子抗体上的Tm增强突变,证明了本文的工作对抗体工程工作的泛化性。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢