
尽管穿膜蛋白(TMP)对分子生物学和医学有着巨大的重要性,但与非TMP相比,TMP的实验三维结构仍较为缺乏 。今天的顶级方法可以准确预测许多结构,但跨膜区域的注释仍然是整个蛋白质组预测的一个限制性步骤。本文提出了一种新的方法,称为TMbed,它从预训练蛋白质语言模型(ProtT5)中输入嵌入,在几小时内就能在一台消费级台式机上完成对整个蛋白质组的α螺旋和β桶状TMP的预测,其性能水平与使用进化信息的方法类似或更好。在每个蛋白质水平上,TMbed正确识别了非冗余数据集中65个β-桶状TMP中的61个(94±7%)和593个α-螺旋状TMP中的579个(98±1%),假阳性率远低于1%(在5859个非膜蛋白中错了31个)。在每段水平上,TMbed平均将10个跨膜区域中的9个正确地置于实验验证的五个残基内。尽管受到GPU内存的限制,本文的方法可以在普通台式电脑使用的标准GPU(如NVIDIA GeForce RTX 3060)上处理多达4200个残基的序列。简而言之,TMbed准确地预测了α螺旋和β桶TMPs,利用蛋白质语言模型和GPU加速,它可以在不到一个小时内预测人类蛋白。
关于数据,本文从OPM收集了所有的α螺旋和β桶状跨膜蛋白的结构文件,并使用SIFTS将它们的PDB链ID映射到UniProtKB蛋白序列。本文放弃了所有嵌合的PDB链和模型结构,以及本文无法映射任何跨膜段的起始或结束位置的PDB链,由此产生了2,053条和206条序列独特的PDB链,分别用于α螺旋和β桶状TMPs。本文使用OPM文件中的ATOM坐标来指定不在膜内的序列段的内/外侧方向。本文手工检查了不一致的注释(例如,如果一个跨膜段的两端有相同的内/外方向),并与PDBTM、PDB和UniProtKB交叉判断,然后要么纠正这种不一致的注释,要么丢弃整个序列。由于OPM不包括信号肽注释,本文将本文的TMP数据集与SignalP 6.0使用的数据集以及UniProtKB/Swiss-Prot中所有具有实验注释的信号肽的序列使用CD-HIT进行比较。对于任何具有至少95%全局序列同一性的匹配序列,将信号肽注释转移到本文的TMPs。本文删除了所有少于50个残基的序列,以避免错误的测序片段产生的噪音,并删除了所有超过15,000个残基的序列,以节省能量(降低计算成本)。最后,本文用MMseqs2将两个TMP数据集中的冗余序列聚类到最多20%的局部成对序列同一性(PIDE)和40%的最小比对覆盖率。最后的非冗余TMP数据集分别包含593个α螺旋TMP和65个β桶状TMP。
关于嵌入,本文用蛋白质语言模型(pLMs)为本文的数据集生成嵌入,使用的是基于Transformer的pLM ProtT5-XL-U50(ProtT5)。本文丢弃了ProtT5的解码器部分,只保留了编码器以提高效率(编码器嵌入的信息量更大),编码器模型将蛋白质序列转换为一个嵌入矩阵,该嵌入矩阵通过一个包含全局和局部上下文信息的1024维向量代表蛋白质中的每个残基,即序列中的每个位置。本文将ProtT5编码器从32位转换成16位浮点格式,以减少GPU上的内存占用。本文采用预训练好的Prot5模型,没有进一步针对具体任务进行微调。本文选择Prot5而不是其他嵌入模型,如ESM-1b(31),是基于本文对该模型的经验和以前项目的比较。此外,Prot5不需要分割长序列,这可能会消除有价值的全局信息,而ESM-1b只能处理多达1022个残基的序列。
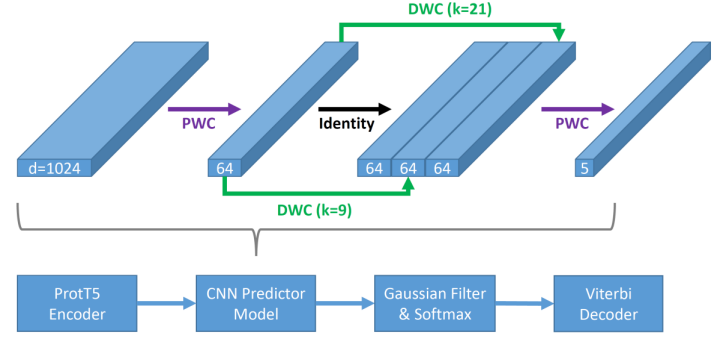
关于模型,如上图所示,TMbed模型由四个部分组成:a)ProtT5编码器将输入序列转换为每个残基的嵌入,序列中的每个残基有1024个维度;b)卷积神经网络(CNN)根据这些嵌入预测类分数;c)高斯滤波器平滑类分数并通过softmax函数将其转换为类概率;d)维特比解码器将类标签分配给序列中每个残基。CNN由四层组成:两个点状卷积(PWC)和两个深度卷积(DWC;核大小为9和21)。第一个PWC和两个DWC的输出也通过层的正常化和ReLU激活函数。
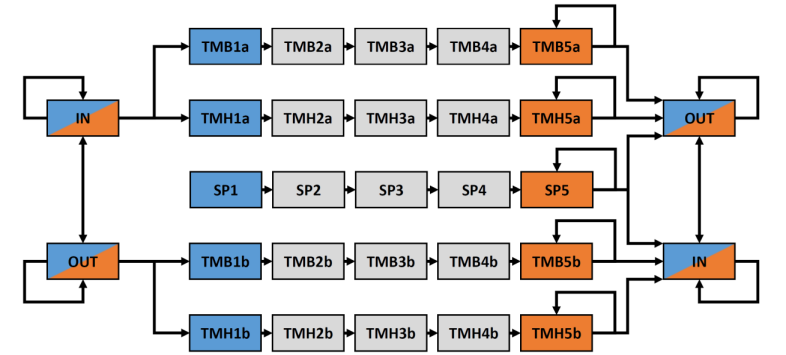
上图展示了维特比解码器中的编码,从一个状态到另一个状态的过渡。本文将跨膜β链(TMB)、螺旋(TMH)和信号肽(SP)分成子状态,以强制执行5个残基的最小片段长度。一个解码的序列必须以蓝色状态之一开始,并且只能以橙色状态之一结束。两侧的IN和OUT状态代表相同的两个内部状态,只是为了简化图形而重复了。
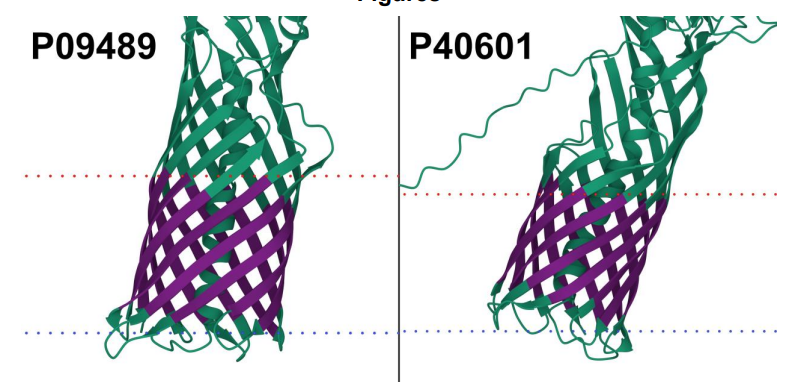
上图展示了跨膜蛋白的示例。细胞外丝氨酸蛋白酶(P09489)和脂肪酶1(P40601)的AlphaFold2结构。TMbed预测的跨膜段(深紫色)与PPM网络服务器预测的膜边界(虚线:红色=外侧,蓝色=内侧)有良好的相关性。使用Mol Viewer创建的图像。尽管本文的数据集将它们列为球状蛋白,但预测的结构表明有跨膜结构域,这与本文的方法预测的片段相一致。预测的结构域与UniProtKB自动注释系统检测到的自动转运器结构域重叠。
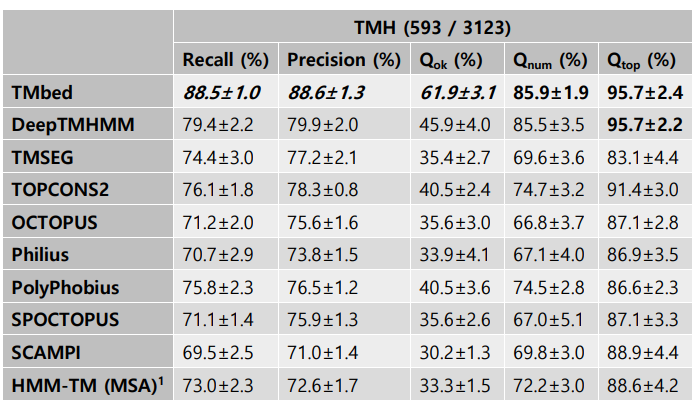
上图展示了593个α螺旋TMP的跨膜螺旋(TMH)预测性能。在所有被评估的方法中,TMbed在跨膜段方面达到了最高的性能。TMbed对TMHs的召回率和精度值为89±1%,明显优于第二好的也是唯一的基于嵌入的方法DeepTMHMM(79±2%,表2)。TMbed基本上预测了几乎62%的跨膜螺旋(TMH)TMPs完全正确(Qok,即所有TMHs在真实注释的±5个残基内)。DeepTMHMM达到第二名,Qok为46±4%。TMbed和DeepTMHMM之间的这一差异是DeepTMHMM和基于进化信息的两种表现第三好的方法TOPCONS2和PolyPhobius的两倍以上。
创新点
- TMbed对α螺旋型(TMH)和β桶型(TMB)跨膜蛋白(TMP)的预测具有很高的准确性,其表现至少与最先进的(SOTA)方法相当,甚至更好,这些方法依赖于来自多序列比对的进化信息。相比之下,TMbed只输入来自蛋白质语言模型(pLM)ProtT5的序列嵌入。
- 本文的新方法特别突出的是它的低假阳性率(FPR),错误地预测不到1%的球状蛋白是TMPs。在正确预测跨膜段方面,TMbed在数字上也超过了所有其他测试方法。
- 尽管TMbed的性能一流,但它更重要的优势是速度:ProtT5编码器的高吞吐率使整个蛋白质组的预测在一个小时内完成。最重要的是,该方法可以在消费级的GPU上运行,就像在最近的游戏和台式电脑中发现的那样。因此,TMbed可以作为一个蛋白质组规模的过滤步骤来扫描跨膜蛋白。
- 用AlphaFold2结构和PPM方法来验证预测的片段,可以结合成一个快速的流程来发现新的膜蛋白,本文已经用几个蛋白质证明了这一点。(参考:Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM
web server: resources for positioning of proteins in membranes. Nucleic Acids Res.
2012;40(Database issue):D370-6) - 最后,本文通过GitHub仓库提供了来自UniProtKB/Swiss-Prot的566,976个蛋白质的预测结果
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢