
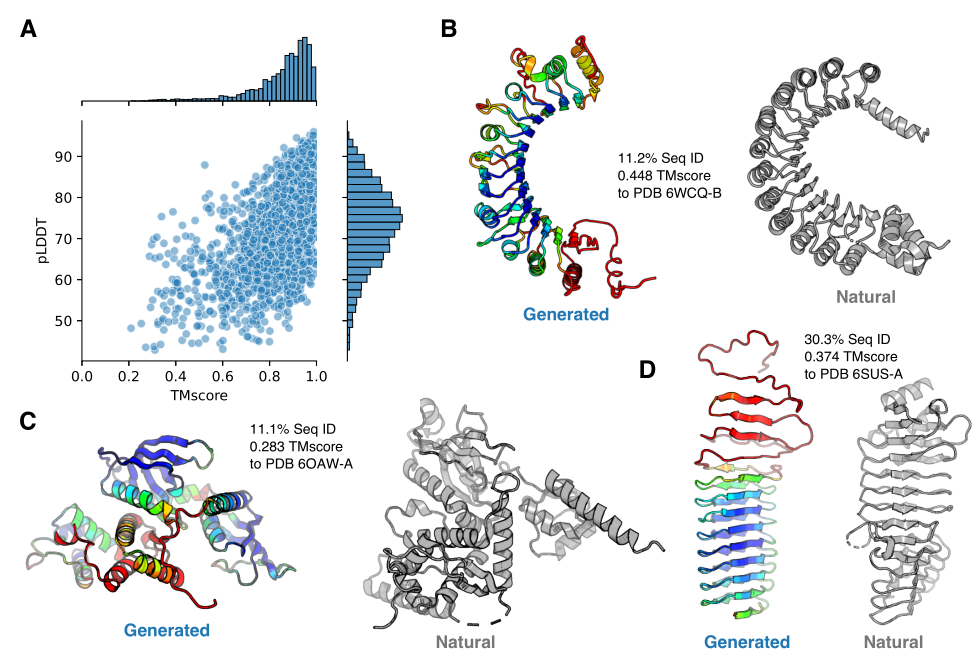
由PLMs生成的序列可以采用各种各样的fold,通常与观察到的蛋白质在序列上有很大的偏差。为了评估PROGEN2模型的生成能力,本文用PROGEN 2-xlarge模型生成了5,000条序列。使用AlphaFold2预测每个序列的三维结构。对于每个结构,本文使用Foldseek确定了结构最相似的天然蛋白质。上图A显示了与天然蛋白的结构相似性TMscore和AlphaFold2预测置信度pLDDT之间的关系。大多数结构都是有信心预测的(TMscore中位数为0.89),并且在PDB中有结构同源物(pLDDT中位数为73.7)。
对预测结构的仔细检查显示了生成序列的几个独特的特征。B图展示了一个采用螺线型折叠的生成序列。PDB中最接近的结构同源物是F-box/LRR-repeat蛋白(PDB ID 6WCQ-B),一个类似折叠的螺线体蛋白。有趣的是,尽管生成的螺线管折叠的内面完全由β片组成(就像在天然蛋白中一样),但外面同时结合了α螺旋和β片,导致更大的中心角和更宽的曲率。此外,尽管采用了类似的折叠,但生成的蛋白质和天然蛋白质之间的序列同一性只有11.2%。
C图展示了一个采用多域α+β-折叠,在PDB中没有发现结构相似的蛋白质。最相似的天然蛋白是一个未定性的蛋白(PDB ID 6OAW-A),TMscore低至0.283,序列重叠度很小(11.1%的同一性)。
D图蛋白的预测结构类似于腺苷酸环化酶毒素的RTX结构(PDB ID 6SUS-A)。这两个结构都采用了类似的β-roll折叠(TMscore 0.374),并且具有中等程度的序列相似性(30.3%)。综合来看,这些例子说明了由PROGEN2生成的序列的一些独特性质,生成的蛋白质类似于天然蛋白质的理想化版本,具有扩展的β片和缩短的连接loop。
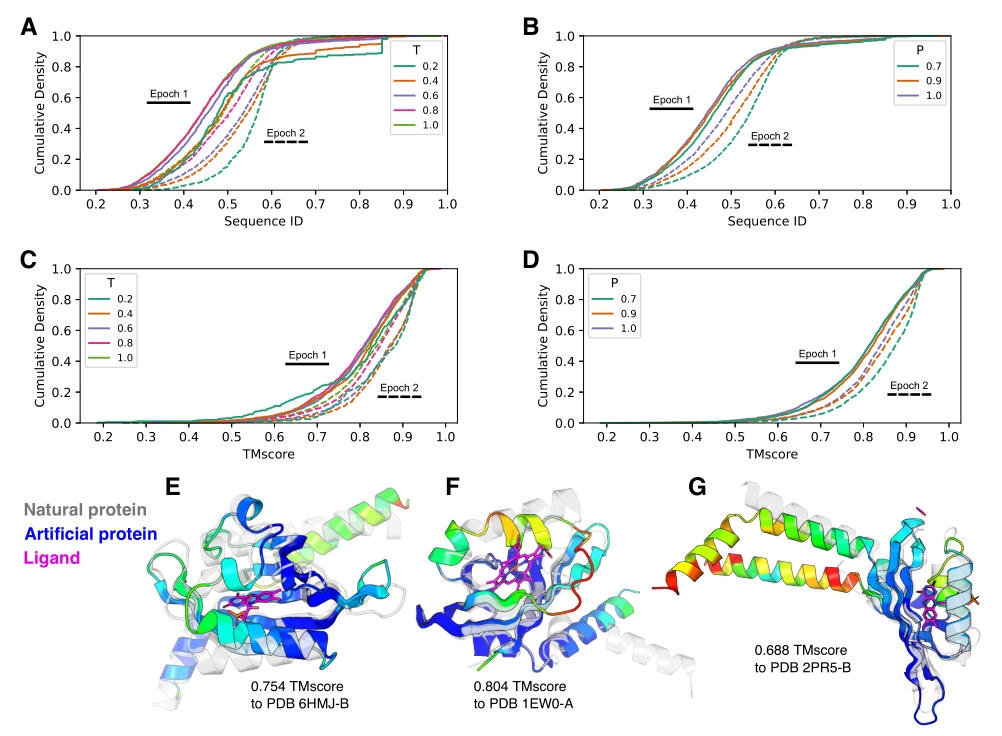
本文在实验中对PROGEN2-large模型在100万个序列上进行了两个epoch的微调,这些序列来自Gene3D和CATH。为了了解微调的效果,本文在微调的第一个和第二个epoch后使用模型参数生成了10,000个序列。对于所有生成的序列使用MMseqs2计算了与训练数据集的序列同一性。为了评估采样参数对共同结构内结构多样性的影响,本文用AlphaFold2预测了所有20000个序列的结构,并用Foldseek计算了针对PDB的TMscores。
图A到D展示了微调的结果,可以看到微调导致生成与训练有更高的序列同一性。另外也可以发现采样参数与序列的新颖性密切相关,即较高的采样温度T或核概率P产生较低的序列同一性,限制性更强的采样参数通常会产生与天然蛋白质更接近的结构。
图E到G显示在较为新颖的结构中,多样性的主要来源是配体结合区,而非结合区则与天然蛋白质相似。图E-F展示了AlphaFold2对配体结合区的预测置信不足,与最接近的天然同源物相比,配体结合区有重排现象。有趣的是,在这两种情况下,预测的结构呈现出适合配体的明显空隙,甚至模仿了天然蛋白质的二级结构。在图G下,生成序列的预测结构较好地地再现了配体结合区域。这些结果表明,由微调模型生成的序列在功能区采样具备多样性,同时保持训练数据集的相似结构。
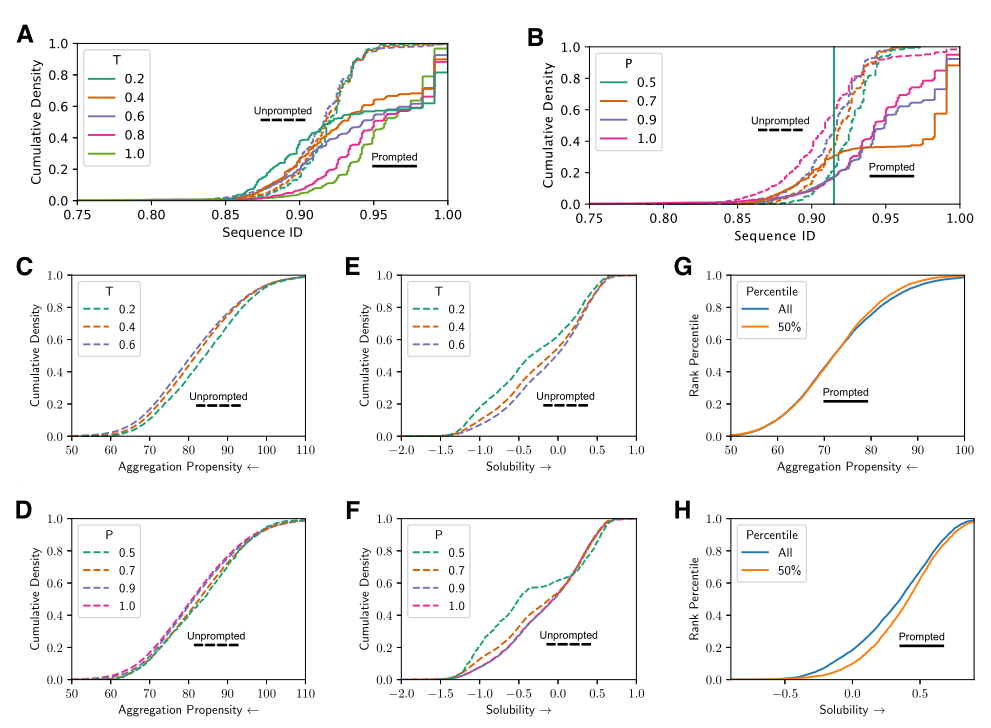
在对来自OAS的可变区序列进行预训练后,使用PROGEN2-OAS模型生成抗体序列,本文包括有初始残基prompt和无初始残基prompt两种生成模式。无prompt模式使用温度(T∈{0.2,0.4,0.6})和核采样概率(P∈{0.5,0.7,0.9,1.0})的笛卡尔乘积的采样参数生成了52K的序列。有prompt的是通过初始化序列与人类重链抗体序列中常见的三残基motif(EVQ),使用温度(T∈{0.2,0.4,0.6,0.8,1.0})和细胞核采样(P∈{0.5,0.7,0.9,1.0})参数的笛卡尔乘积,同样产生的47万全长序列。用MMseqs2计算生成的序列与训练数据集的序列同一性。IgFold被用来预测所有生成的抗体序列的结构,PyRosetta对模式输出进行了refine。
在图A-B中,本文比较了无prompt的和prompt的世代与训练分布的序列相似性。值得注意的是,prompt序列与训练分布的序列同一性明显提高,这可能是由于包含了高度保守的FW1区域,而该区域在N端截断的无prompt序列中经常缺失。另外可以观察到更严格的采样参数(更低的温度,更高的核概率)与训练数据集的序列同一性之间的反比关系。
潜在的抗体研发往往需要广泛的优化以改善其物理特性。这些特性统称为可开发性,包括热稳定性、表达能力、聚集倾向和可溶性。本文专注于根据其SAP评分和CamSol-intrinsic评价生成序列的聚集倾向和溶解性。图C-F显示对于聚集倾向和溶解度,用较少的限制性参数生成的序列显示出更好的可开发性。
鉴于PLMs有效的零样本预测能力,作者表示还研究了是否可以用一个统一的预训练模型来过滤生成的抗体库并改善其可开发性。图G-H中比较了全部生成序列与PROGEN 2-base模型评分的前50%的聚集倾向和溶解度。在排名靠前的序列中,聚集倾向只略有改善,而序列的溶解度则显示出有利的转变。这些结果为用PLMs生成抗体序列库提供了有意义的指导。在实践中,用限制性较小的采样参数生成和用通用PLM过滤应该提供最可开发的序列集。

为了与Rita模型进行适当的比较,其结构与PROGEN2相似,但在不同的数据分布上进行了训练,本文首先对DeepSequence的适应度景观的零样本性能进行了表征,其主要由单替换的深度突变扫描实验组成。从上图发现,最小的模型(PROGEN2-small),其参数比RITA-XL少一个数量级,在样本任务中表现出更高的平均性能,表明预训练数据分布的重要性。与RITA相比,PROGEN2的训练数据包罗万象,由Uniref的同一度筛选序列和元基因组来源的序列组成。最好的PROGEN2模型优于或匹配所有其他基线,显示了模型训练的序列集的重要性。

本文所比较的大多数可用的适应度试验都集中在具有大量进化相似序列的研究良好的蛋白质上,并测量与野生型序列只有一到两个突变的突变体的适应度/功能。从经验上看,本文发现在序列缺乏相似同源序列时,较大的模型在适应度估计方面优于较小的模型,如上图展示的一样。这点对于GB1尤甚,这是一个具有挑战性的低同源性蛋白质,在具有非线性外显性的位置上发生了突变,最大的模型xlarge在零样本识别最高适应度变体时可能表现出突然的性能提升,即所谓的大型语言模型的突发(emergent)能力:一种能力在较小的模型中不存在,但在较大的模型中存在,则这个能力是emergent的。

在抗体特异性方面,本文的结果再次表明需要更加关注训练期间提供给模型的序列的分布。上图展示了抗体的亲和力(KD)和一般属性(表达量和熔化温度TM)的零样本预测。来自免疫组库测序研究的样本似乎是一个直观的选择,用于学习对抗体适应度预测任务有用的强大表征。有趣的是,使用抗体训练的PROGEN2-OAS模型与在通用蛋白质数据库上训练的预训练模型xlarge相比表现不佳。尽管相应的抗原没有提供给模型进行似然计算,但结合预测的性能是不可忽略的,在实际的抗体工程活动中可能是有用的。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢