
一旦经过训练,DEDAL就会产生专门为每一对新的序列计算的间隙和替换的评分矩阵。此外,间隙和替换的分数是有背景的:对于每一对位置,它们取决于要对齐的完整序列。然后用一个标准的SW算法使用这些参数计算出最佳的排列。本文表明,DEDAL可以在带有加速器的现代硬件上有效地训练。一旦训练完成,DEDAL与标准SW相比,为远程同源物预测的对准质量提高了2-3倍,并产生了一个能更准确检测远程同源物的对准分数。
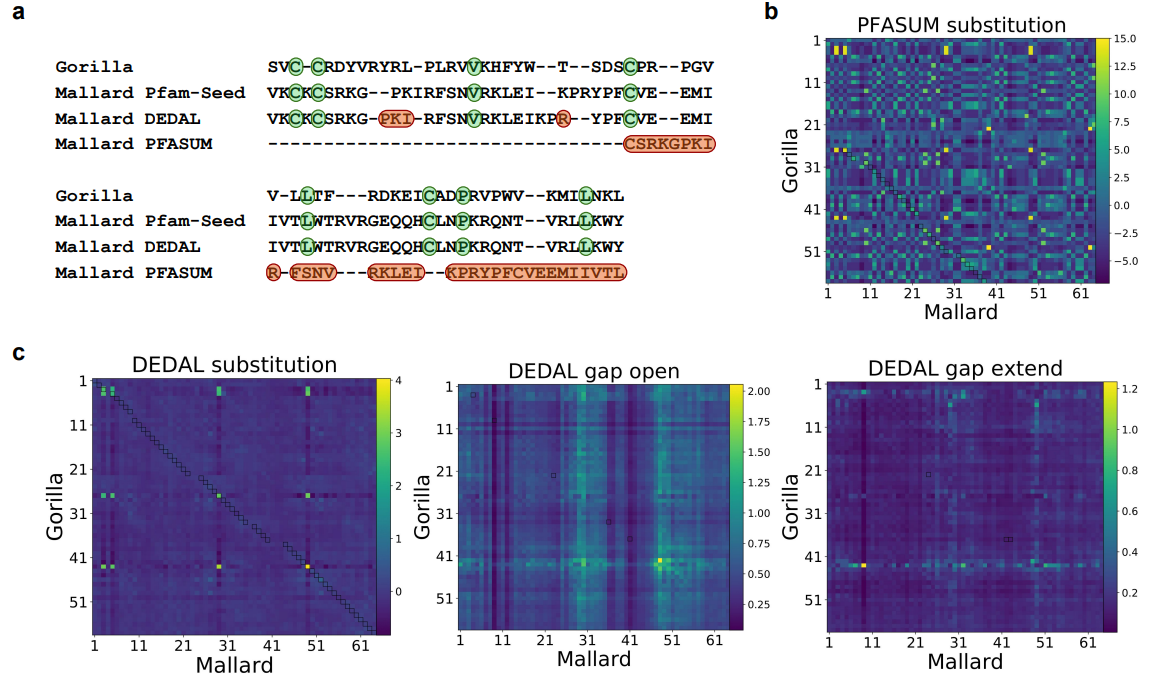
上图展示了来自Pfam-A种子的两个蛋白域序列比对例子。
a. 分别从Pfam-A种子数据库(第二行)、DEDAL预测(第三行)和用PFASUM70替代矩阵预测(第四行)进行的比对。本文显示了Pfam-A种子和DEDAL对准的两个序列中的所有残基,但没有显示PFASUM的序列中对准的上游和下游的未对准残基。绿色突出显示的残基对应于正确对齐的保守残基,而红色显示的残基对应于预测对齐和Pfam-A种子对齐之间的差异。
b. 来自PFASUM替代矩阵的所有残基对之间的替代分数。
c. 由DEDAL预测的SW参数。
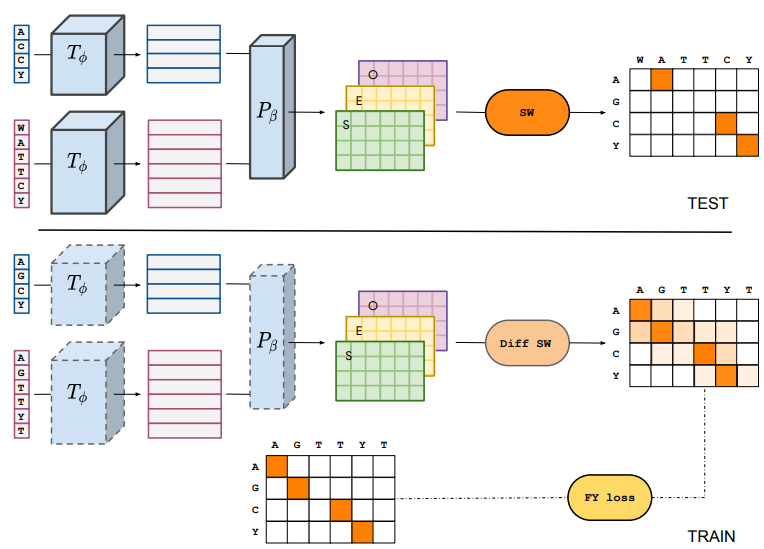
在技术方面,本文探索了两种方法来创建一个可区分的SW对齐模块,需要在 "学习对齐 "任务中训练DEDAL的参数,使用平滑技术或扰动技术;本文发现两者在性能上没有明显区别,并在最终的DEDAL模型中实施了基于扰动的方法。关于用于训练DEDAL的排列组合,本文发现,当本文希望DEDAL能够预测准确的局部排列时,使用Pfam扩展域而不是Pfam域是有益的。在遮蔽语言建模任务中预训练DEDAL时,将与分布外家族相关的序列从 "蛋白质宇宙 "中排除,导致远程同源物的性能略有下降,尽管相对于与基线的性能差距来说并不明显。
关于端到端联合训练变换器和参数器的策略,本文发现这确实明显优于更经典的两步策略,即首先在屏蔽的语言建模任务中训练变换器编码器,然后通过保持变换器固定在 "学习对齐 "任务中训练参数器。这表明,一个通用的语言模型,如ESM,是不够的,至少应该进行微调,以达到对齐的最佳性能。
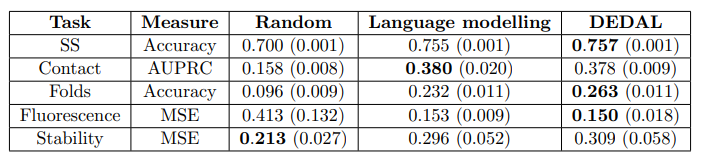
上图展示了学习的嵌入在下游任务的应用情况。本文通过简单地训练一个模型来评估与上下文相关的嵌入的好处,在这个模型中,替换成本被限制为只取决于要对齐的氨基酸;不难看出,本文观察到这个模型的性能有很大的下降,达到了与 "对准 "中表现最好的替换矩阵差不多的性能。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢