
Language models of protein sequences at the scale of evolution enable accurate structure prediction
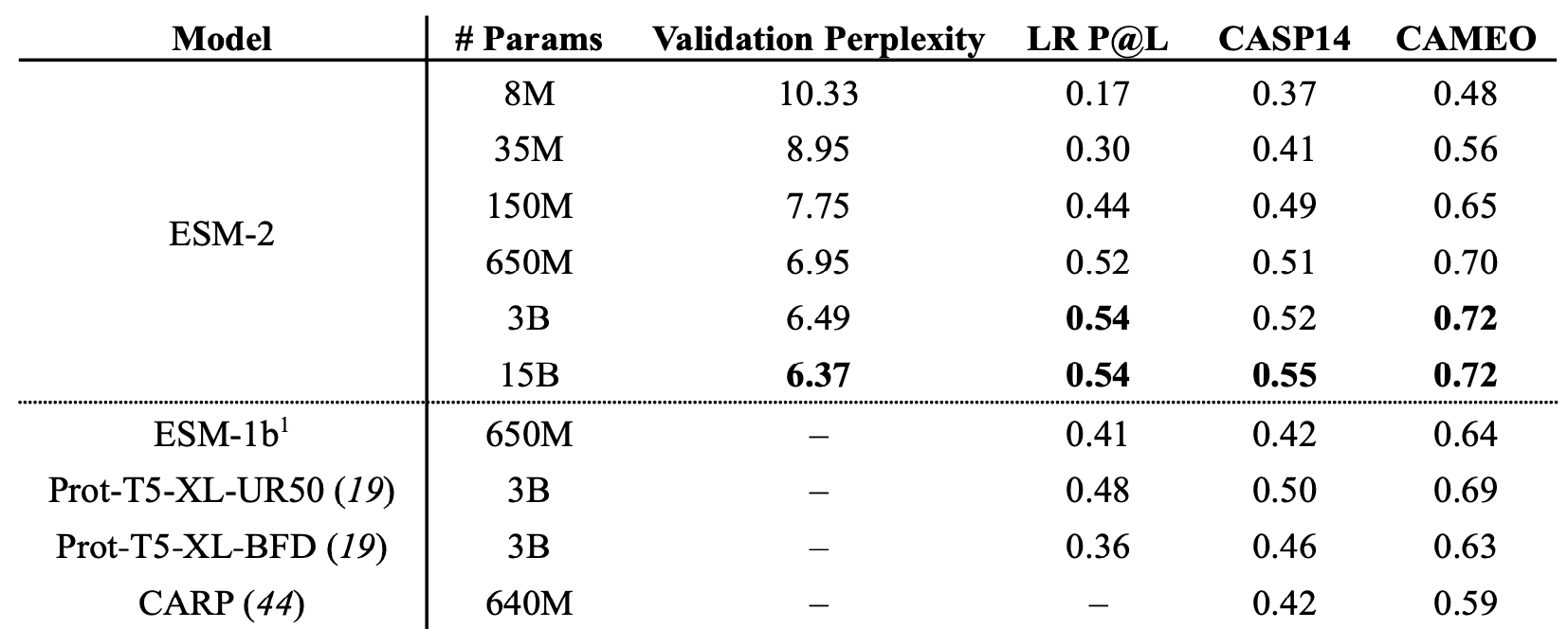
各个预训练模型在困惑度,接触预测等任务上的性能比较。
esm2是在Uniref数据库上训练的一系列transformer encoder模型,参数量最多15B,位置嵌入使用了RoPE(Rotary Position Embedding)以允许模型推断到它所训练的上下文窗口之外。
可以发现相对尽管训练时间较短,15B参数的ESM-2模型在CASP14上具有最低的验证复杂度和最高的TM分数。150M参数的ESM-2模型在基于结构的任务上优于650M参数的ESM-1b模型,而650M参数的ESM-2模型与3B参数的Prot-T5-XL-UniRef50模型相当,这表明ESM-2模型的参数效率高得多。
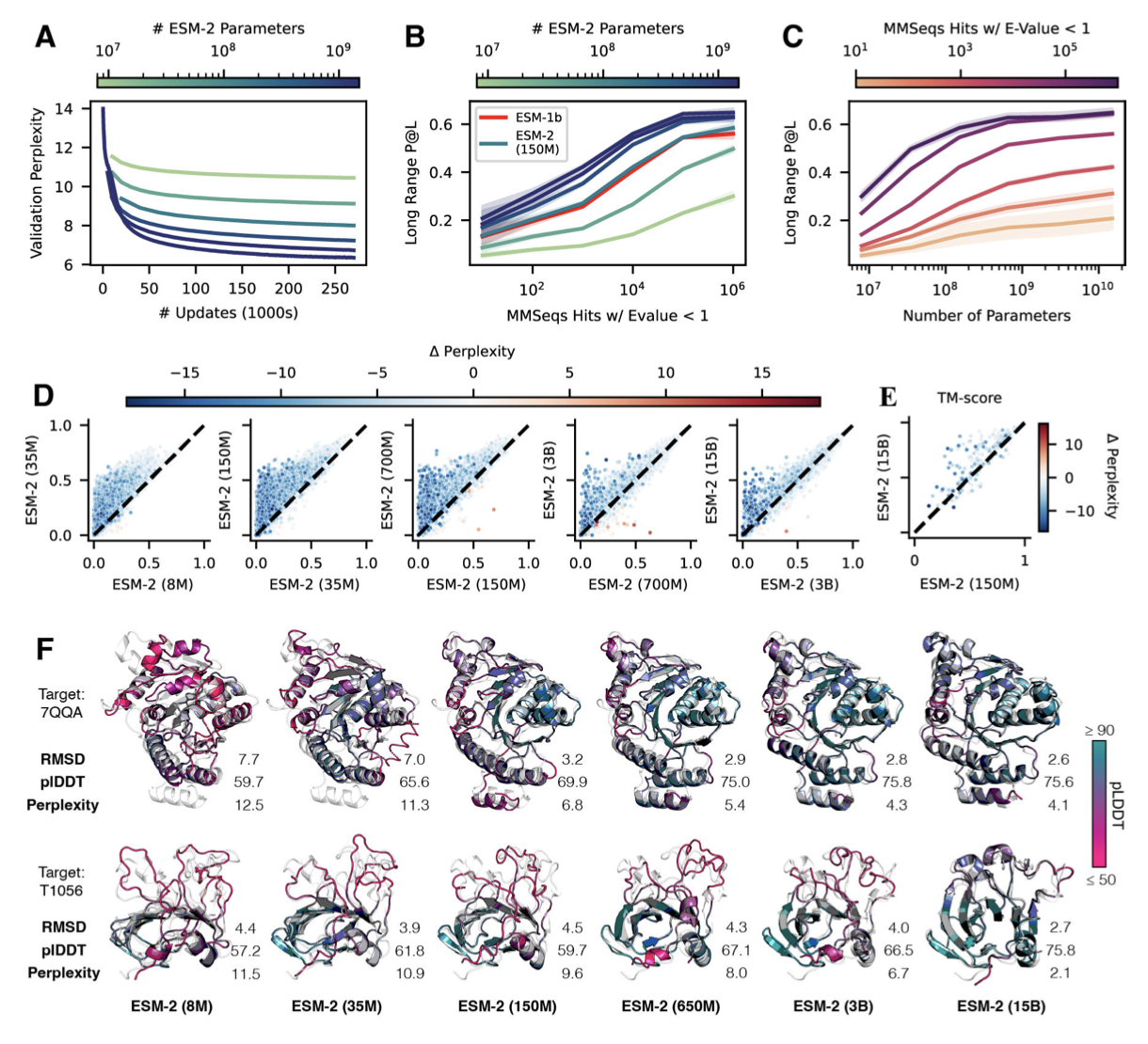 (A) 所有规模的ESM-2模型通过27万次更新的训练曲线。
(A) 所有规模的ESM-2模型通过27万次更新的训练曲线。
(B)ESM-2模型不同规模的无监督接触预测性能,本图展示了性能与搜索训练集时的MMseqs hits数的关系,较大的模型在所有级别都表现较好,150M参数的ESM-2模型与650M参数的ESM-1b模型的表现相当。
(C) 本图展示了随着模型规模的增加,对具有不同数量MMseqs hits的序列的相关性变化,对于具有O(10^4) MMseqs hits率的序列,改进幅度最大。
(D) 从左到右显示了从8M到15B参数的模型,在无监督接触精度方面连续比较了较小的模型(X轴)和下一个较大的模型(Y轴)。点对应于PDB蛋白,并以较小模型和较大模型之间序列的困惑度的变化来标示,接触预测性能变化大的序列也表现出以困惑度衡量的语言模型理解的巨大变化。
(E) CASP14和CAMEO测试集的TM score,模型是在150M参数(X轴)和15B参数(Y轴)ESM-2上训练的结构模块。
(F) 在所有ESM-2模型下,对CAMEO结构7QQA和CASP target1056从左到右的结构预测,用pLDDT着色(粉红色=低,茶色=高)。对于7QQA,预测精度在150M参数阈值时突然提高,此后缓慢提高。对于T1056,预测精度在15B参数阈值时突然提高。
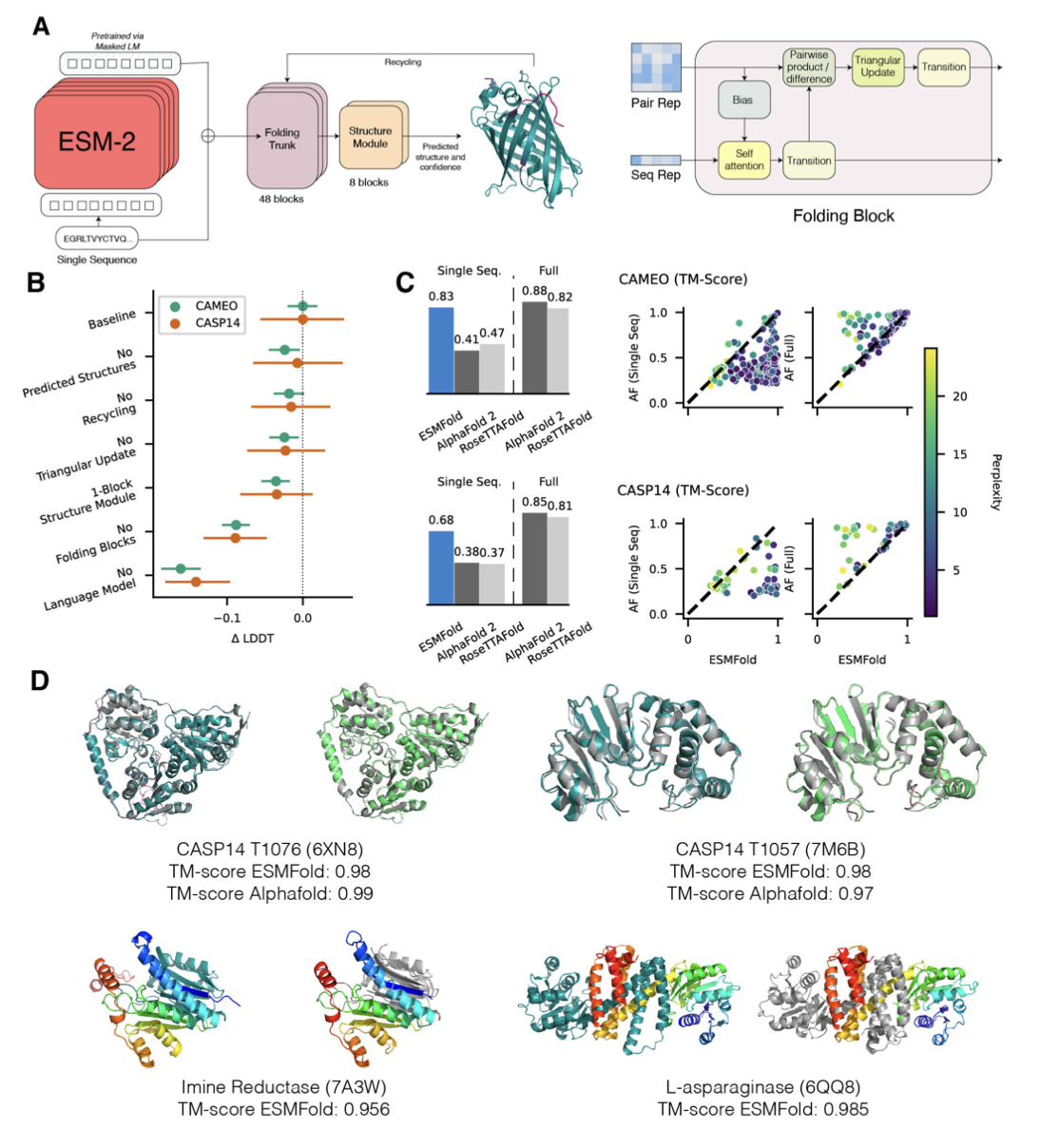
ESMFold能够从单一序列中进行精确的结构预测。
(A) ESMFold模型架构,箭头显示了网络中从语言模型到folding trunk到结构模块的信息流,结构模块输出三维坐标和置信度,folding trunk是AlphaFold2中描述的EvoFormer的简化单序列版本。
(B) 各种消融实验对ESMFold测试性能的影响,语言模型是其中最重要的。
(C) 当给定一个单一的序列作为输入时,ESMFold优于RoseTTAFold和AlphaFold2,即使在CAMEO上给定完整的MSA时,也与RoseTTAFold有竞争力。散点图显示了ESMFold(x轴)对AlphaFold2(y轴)的性能,以困惑度为指标。可以esmfold低困惑度的蛋白质与AlphaFold2的得分相似。
(D) 第一行显示测试集预测的ESMFold为茶色,真实结果为灰色,AlphaFold2的预测为绿色。粉红色显示了ESMFold和AlphaFold2的低预测值lDDT。第二行显示了复合物的预测结果;ESMFold的B链为茶色(左),真实结果为灰色(右);A链按照彩虹的颜色变化,从蓝色(N-末端)到红色(C-末端)着色。
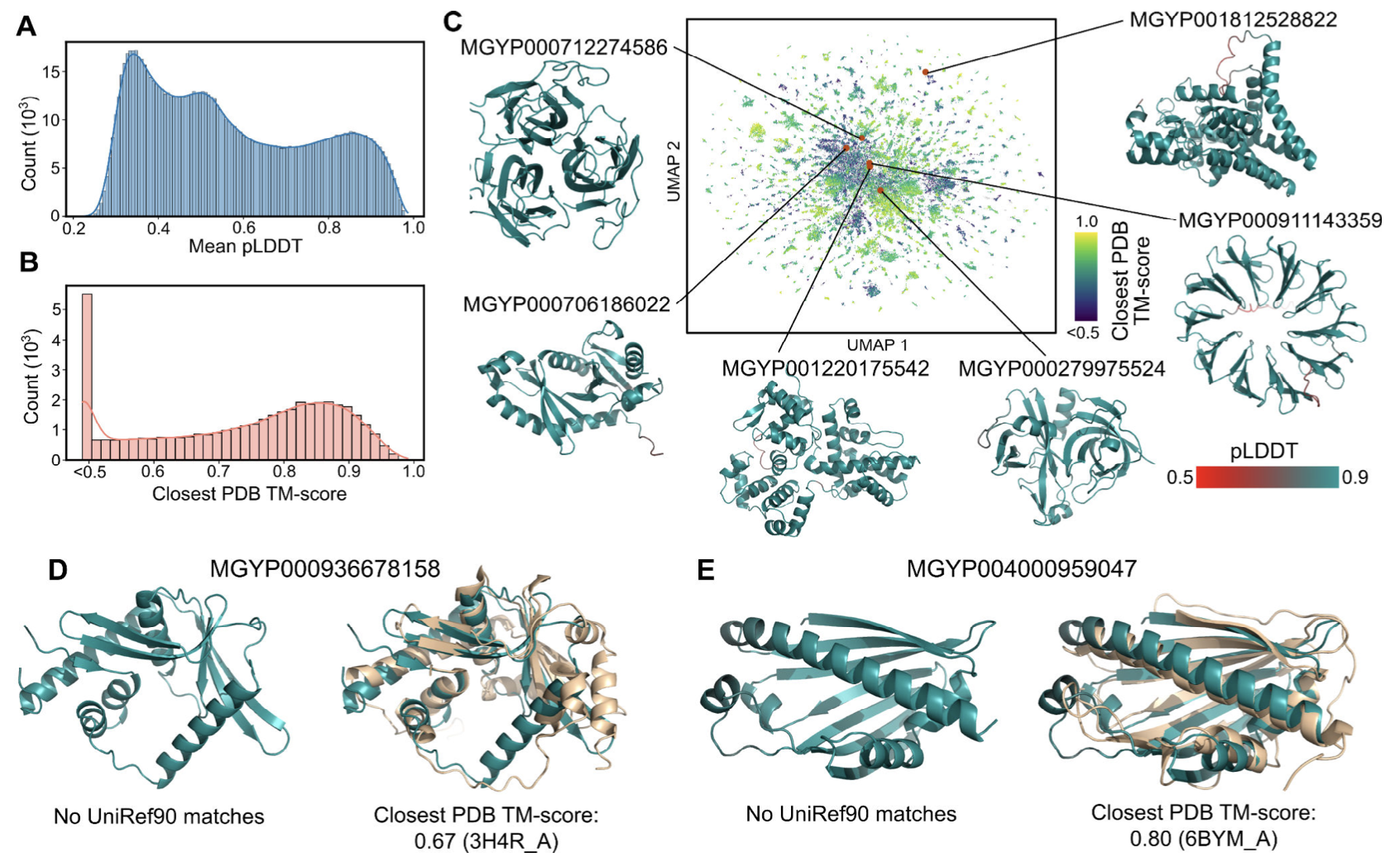
上图展示了探索元基因组结构空间
(A) 来自MGnify数据库的100万个ESMFold预测结构中每个结构的平均pLDDT值的分布。
(B) 在59K个最高置信度(平均pLDDT>0.9)的结构中,每个结构与最相似的PDB结构的TM分数分布。该值是通过Foldseek搜索得到的,该搜索不报告TM分数低于0.5的结构。
(C) 高可信度的蛋白质结构用UMAP算法进行二维可视化,并根据与最近的PDB结构的距离进行着色,其中与已知结构相似度低的区域用深蓝色着色。
(D, E)两个ESMFold预测结构的例子,与PDB中的实验结构有很好的一致性,但与UniRef90中的任何序列都有很低的序列同一性,当序列信息不足时,有可能实现基于结构的功能洞察。D展示了MGYP000936678158的预测结构与细菌核酸酶的实验结构相一致(浅棕色,PDB: 3H4R),E展示了 MGYP004000959047的预测结构与细菌甾醇结合域的实验结构相一致(浅棕色,PDB: 6BYM)。
创新点
- 用无监督学习目标训练的语言模型在一个进化多样的蛋白质序列的大型数据库中能够对蛋白质结构进行原子层次分辨率预测。
- 本文提出了ESM-2,是迄今为止训练的最大的蛋白质语言模型,其参数仅比最近开发的最大文本模型少一个数量级。
- 本文表明,ESMFold的性能的最大驱动力是语言模型。由于语言模型的迷惑性和结构预测的准确性之间有很强的联系,本文发现,当序列被ESM-2很好地理解时,可以获得与最先进的模型相当的预测结果。
- ESMFold获得了准确的原子分辨率结构预测,推理时间比AlphaFold2提高了一个数量级。在实践中,由于ESMFold消除了搜索进化相关序列以构建MSA的需要,因此速度上的优势甚至更大。
- 推理时间的优势使得有效地绘制大型元基因组学序列数据库的结构空间成为可能。除了基于结构的工具来识别远程同源性和保守性,用ESMFold进行快速准确的结构预测有助于在大量新序列集合的结构和功能分析中发挥作用。
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢