【标题】HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
【作者团队】Xiaomin Fang, Fan Wang, Lihang Liu, Jingzhou He, Dayong Lin, Yingfei Xiang, Xiaonan Zhang, Hua Wu, Hui Li, Le Song
【发表时间】2021/07/28
【机 构】百度、百图生科
【论文链接】https://arxiv.org/pdf/2207.13921v1.pdf
【代码链接】 https://github.com/PaddlePaddle/PaddleHelix/tree/ dev/apps/protein_folding/helixfold-single
【工具链接】https://paddlehelix.baidu.com/app/drug/protein-single/forecast
基于人工智能的蛋白质结构预测方法,如AlphaFold2,已经达到了接近实验的准确性。这些先进的方法主要依靠多序列比对(MSA)和模板作为输入,从同源序列中学习共进化信息。然而,从蛋白质数据库中搜索MSA和模板是非常耗时的,通常需要数十分钟。因此,本文试图通过只使用蛋白质的一级序列来探索快速蛋白质结构预测的极限。HelixFold-Single的提出是为了将大规模的蛋白质语言模型与AlphaFold2的几何学习能力结合起来。HelixFold-Single首先利用自监督的学习范式,用数以百万计的序列预训练一个大规模的蛋白质语言模型(PLM),它将作为MSA和模板的替代品来学习共进化信息。然后通过结合预训练的PLM和AlphaFold2的基本组件,得到了一个端到端的仅从主序列预测原子的三维坐标的可微模型。HelixFold-Single在CASP14和CAMEO数据集中得到了验证,在具有大型同源家族的目标上实现了与基于MSA的方法具有竞争能力的准确度。此外,HelixFold-Single比蛋白质结构预测的主流方法消耗的时间要少得多,这表明它在需要大量预测的任务中具有潜在应用。
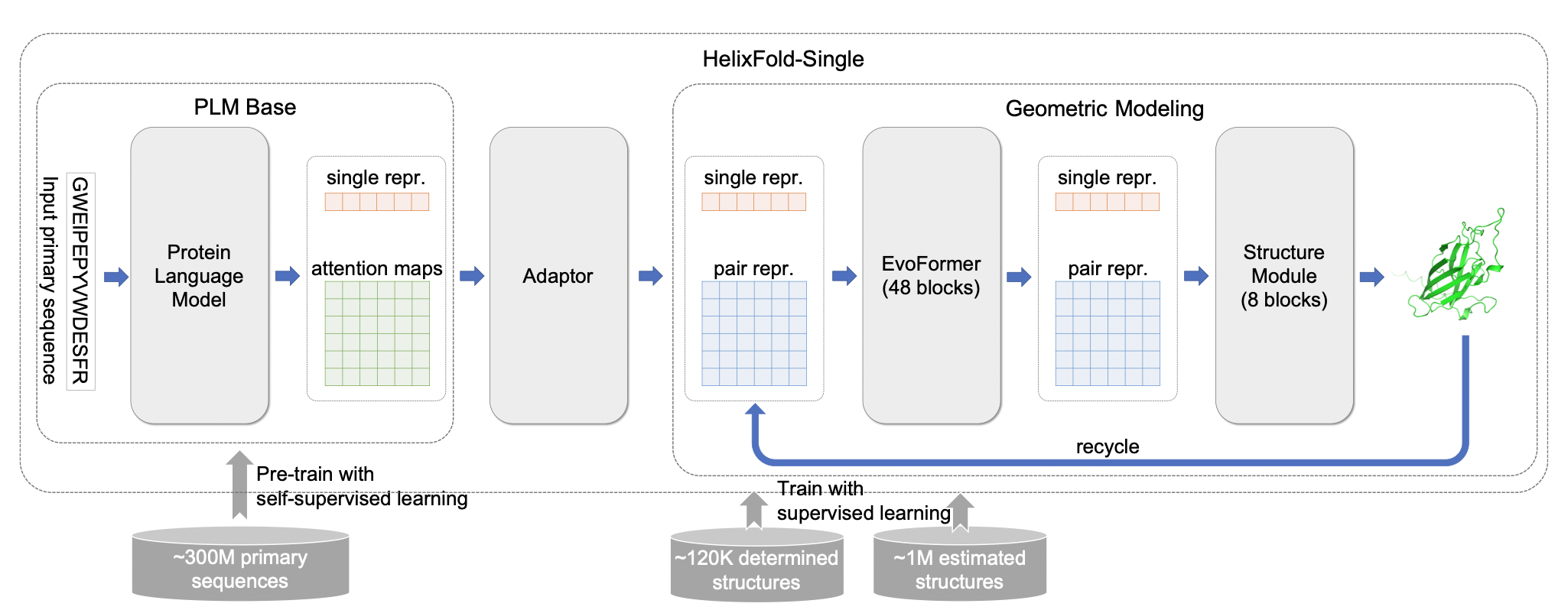
HelixFold-Single由三个组件组成:PLM、适应器和几何模型。一个大规模的预训练语言模型被用来编码参数中的共进化信息,它被用作MSA和模板的替代。然后,在几何模型中,仿照AlphaFold2,本文使用修改过的EvoFormer和结构模块来充分交换序列表征和pair表示之间的信息,以捕捉几何信息并恢复原子的三维坐标。本文采用Adaptor层从PLM中提取协同进化信息,以有效地生成作为几何建模输入的序列和pair表征。整个可微方法是通过自监督预训练和监督学习来训练的,自监督预训练包括大量未标记的主要序列,监督学习包括几何标签。
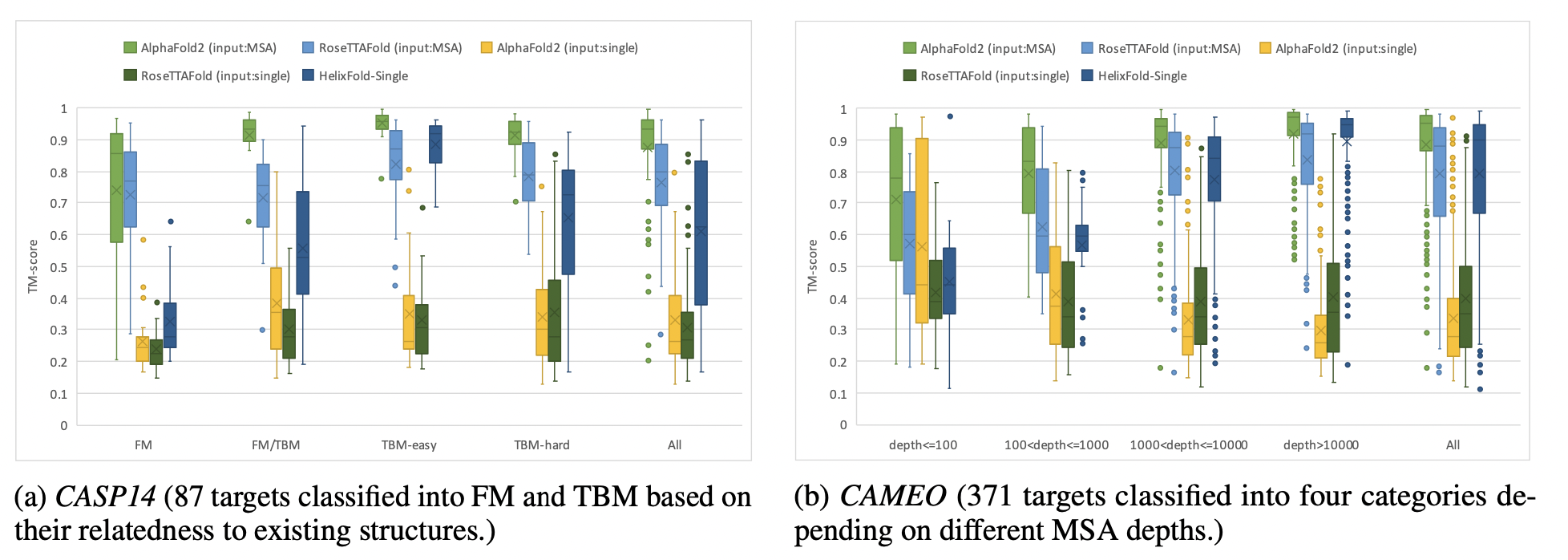
上图展示了Helixfold-Single在CASP14和CAMEO中的表现,可以发现:
- 总的来说,HelixFold-Single在CASP14和CAMEO上大大超过了所有无MSA的方法,在某些情况下与基于MSA的方法具有竞争力。值得注意的是,HelixFold-Single在CAMEO上的准确度与以MSA为输入的AlphaFold2相当,并超过了另一个强大的基线以MSA为输入的RoseTTAFold。HelixFold-Single证明了将PLM纳入几何模型用于蛋白质结构预测的巨大潜力。
- HelixFold-Single可以与基于MSA的方法在具有大量同源家族的靶点上表现相当,例如CASP14中的TBM- easy domain targets,其同源序列的中位数为7个,CAMEO中有超过1000个同源序列(MSA深度>1000)。这些结果表明HelixFold-Single的准确性与同源序列的丰富程度相关,揭示了HelixFold-Single采用的大规模PLM能够嵌入基于MSA的方法所使用的MSA和模板的信息,如共进化知识。
- HelixFold-Single与其他不含MSA的方法相比,在CASP14和CAMEO的所有类别上表现出极大的优越性。由于AlphaFold2和RoseTTAFold在训练过程中依赖MSA和模板作为输入,因此这些方法在只采用单一序列作为输入时,很难提供准确的预测结果。
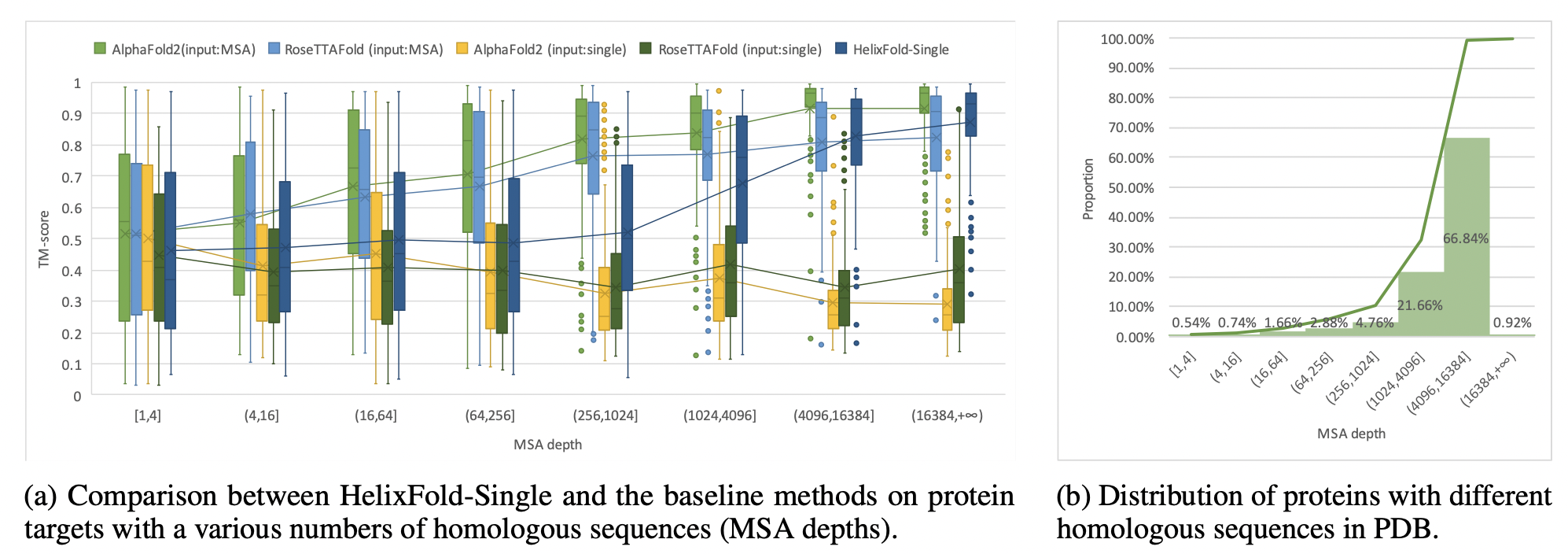
上图展示了HelixFold-Single与同源序列数量的关系。
具体而言作者从PDB收集了2020-05和2021-10年间发布的靶点,从中挑选出同源序列相对稀少的与CASP14和CAMEO的数据混合,作为新的测试集。可以看到,随着可用同源序列的增加,HelixFold-Single和基于MSA的方法的平均TM-score都在增加,而其他无MSA的方法的分数则在下降。在同源序列稀少的蛋白质上,所有比较方法的TM-score都不理想。对于具有较大同源家族的蛋白质,特别是具有数千条以上同源家族的蛋白质,HelixFold-Single可以与基于MSA的方法竞争。鉴于PDB中90%的目标有超过1024个同源序列,本文假设HelixFold-Single在最常研究的蛋白质上可以达到令人满意的精度。
创新点
通过利用大规模预训练模型来嵌入同源信息,本文证明它可以作为MSA和模板的替代品来减少蛋白质结构预测方法所需的时间消耗。
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢