
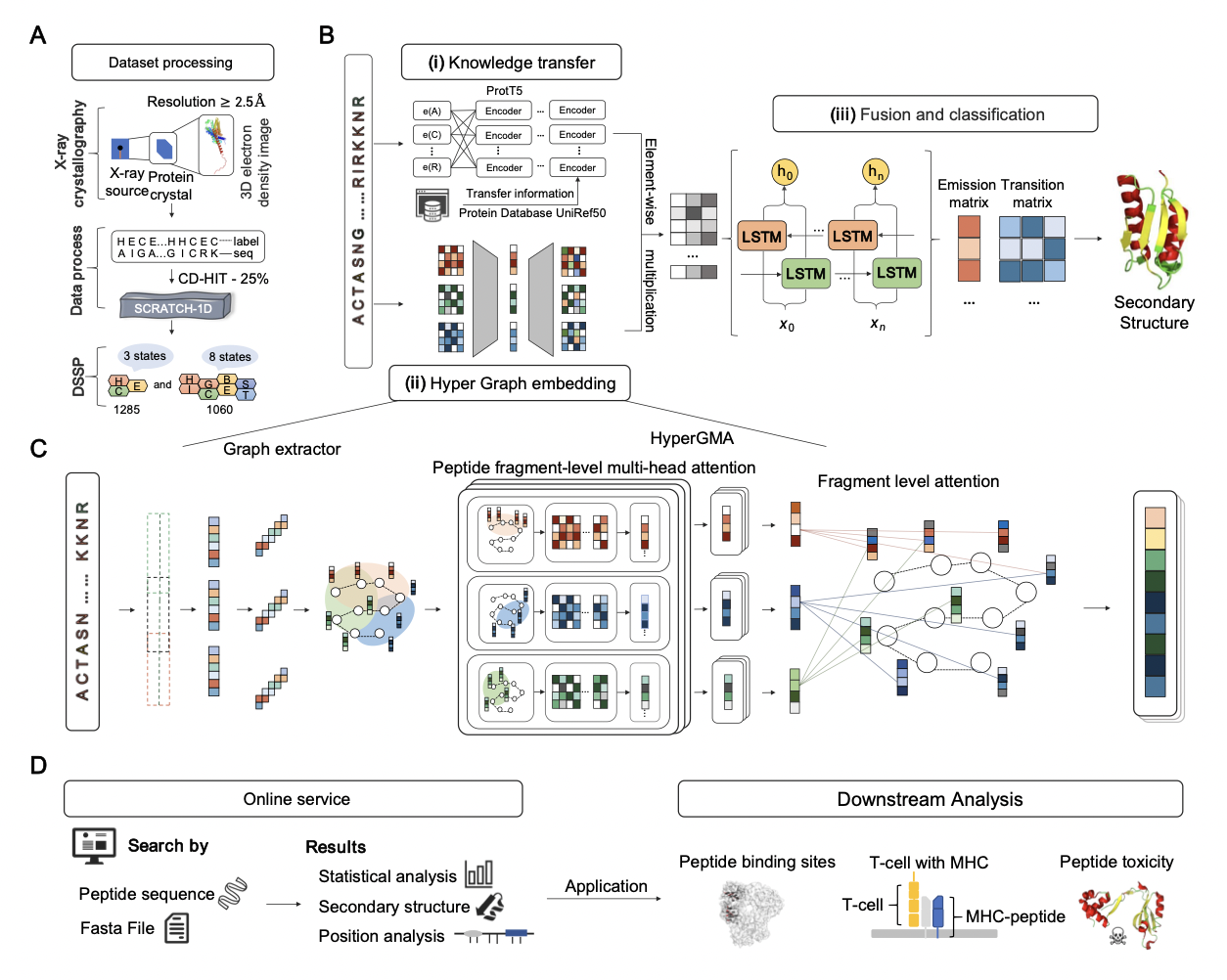
上图展示了PHAT的工作流程和框架。
(A) 数据集处理。本文从SCRATCH-1D中提取了基准数据集,其中蛋白质和肽的结构是用X射线晶体学得出的,并以至少2.5埃的分辨率操作,用于三态和八态二级结构。
(B) PHAT的框架。该框架由三个模块组成:(i)知识迁移模块,(ii)超图嵌入模块,以及(iii)融合和分类模块。在知识迁移模块中,原始序列被预训练的蛋白质模型编码,以获得肽残基的特征。在超图嵌入模块中,肽序列被构建成超图结构并被HyperGMA嵌入。在融合和分类模块中,知识迁移模块和超图嵌入模块的输出首先通过元素相乘进行融合,并通过Bi-LSTM进行更好的整合。然后,Bi-LSTM的输出被输入到CRF层,可以预测相关残基的二级结构。
(C)超图嵌入模块的细节。在图提取器部分,首先将肽序列切成具有特定长度的片段,并构建为超图结构的超边。然后将超边切成残基,作为超图结构中的超节点来构建。接下来,图提取器中的超图结构被输入到HyperGMA中,通过注意力机制捕捉残基和肽片段的多尺度关系。
(D) 在线服务。PHAT的网络服务器可以免费为研究人员提供三态或八态二级结构的肽的细节。统计分析和位置分析。模型的预测结果可以应用于许多下游任务,如下游分析。
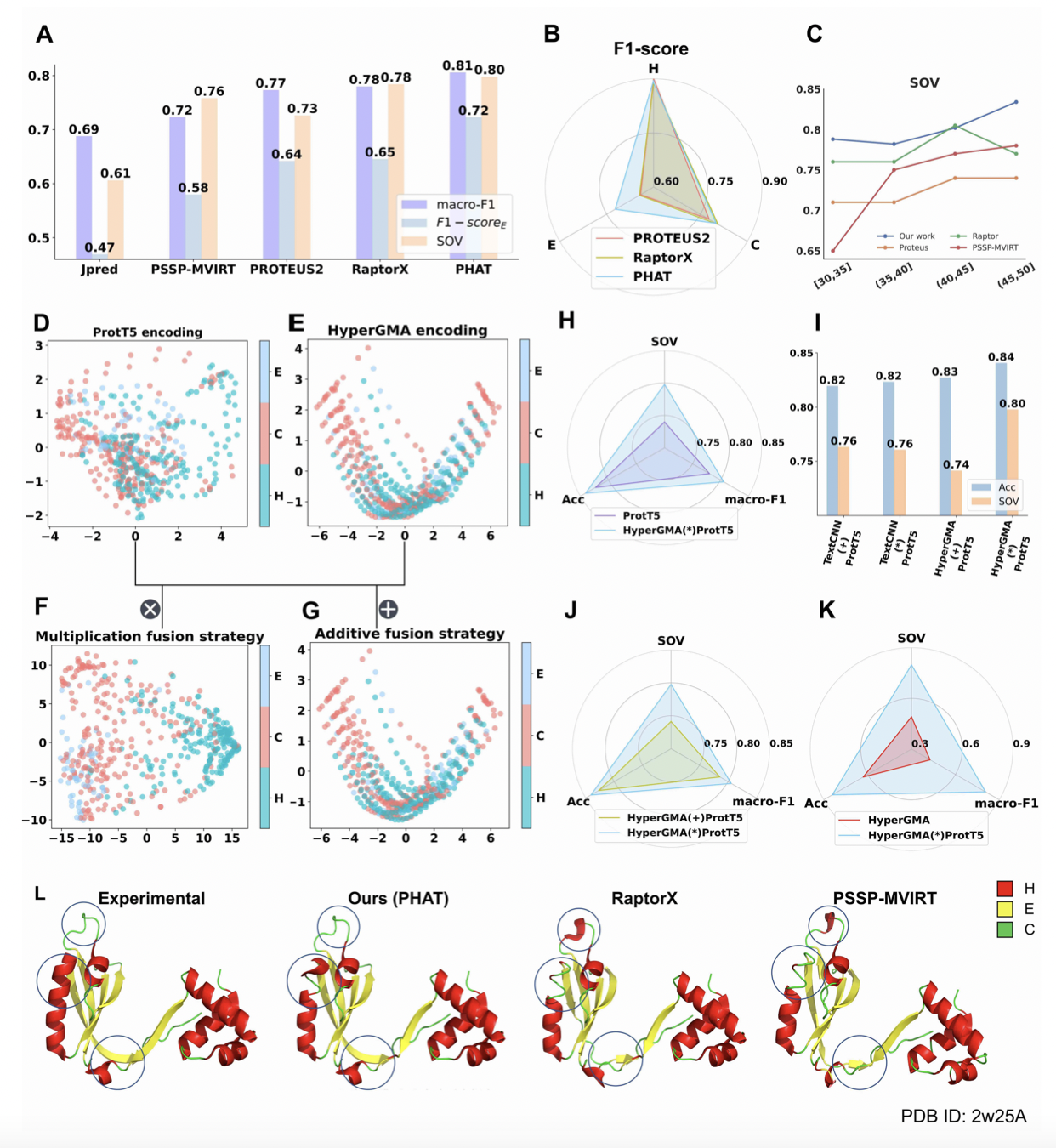
上图展示了本文方法和现有方法在独立测试子集,不同编码策略的比较,以及不同方法在一条肽上的可视化。
(A)以SOV(segment overlap measure)、macro-F1和F1-scoreH作为评价指标的表现。
(B)以三个子结构下的F1-scores作为评价指标的表现。
(C)四种方法在不同长度区间的SOV。
(D-G)表示ProtT5、HyperGMA的单个特征以及乘法或加法融合特征的PCA可视化结果。
(H, J, K)表示乘法融合策略与其他三种策略的比较。
(I)表示HyperGMA和TextCNN的性能比较。
(L) 本文的方法和其他两种方法对PDB ID: 2w25的肽的预测结果的可视化。
创新点
- 本文的方法可以通过超图多头注意网络捕捉到更多的肽序列的上下文信息,从而可以在局部连续的序列区域做出更正确的预测,对两个肽(PDB ID:2w25A和1ejbA)的预测的可视化证明了这一点。
- 本文方法除了能够捕获上下文信息外,还可以通过使用ProtT5预训练模型获得肽序列的长期和生物语义知识,从而达到良好的预测性能。用于二级结构预测的肽长偏好实验表明,尽管被测方法的预测性能随着序列长度的下降而下降,但我们的方法在分析较短的肽序列时取得了比其他现有方法更好的性能。这表明本文模型可以整合上下文信息和知识来进行预测。
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢