
【论文标题】PeTriBERT : Augmenting BERT with tridimensional encoding for inverse protein folding and design
【作者团队】Baldwin Dumortier, Antoine Liutkus, Clément Carré, Gabriel Krouk
【发表时间】2022/08/13
【机 构】蒙彼利埃大学、BionomeeX
【论文链接】https://doi.org/10.1101/2022.08.10.503344
蛋白质研究是生物学的重点。自从最近新的折叠方法取得突破以来,可用的结构数据量不断增加,这缩小了基于数据驱动的序列方法和结构方法之间的差距。本文专注于逆折叠问题,从蛋白质三维结构中预测条氨基酸序列。为此,作者从自然语言处理增强的三维结构数据中引入了一个简单的Transformer模型,并把由此产生的模型称为PeTriBERT:在BERT模型中嵌入三维表征的蛋白质。本文在从新获得的AlphaFoldDB数据库中检索到的350,000多条蛋白质序列上训练这个小型的4000万参数模型。使用PetriBert,能够在与吉布斯采样结合的虚拟计算中生成具有类似GFP结构的全新蛋白质,这表明PetriBert确实捕捉到了蛋白质的折叠规则,并可能成为蛋白质设计的一个重要工具。
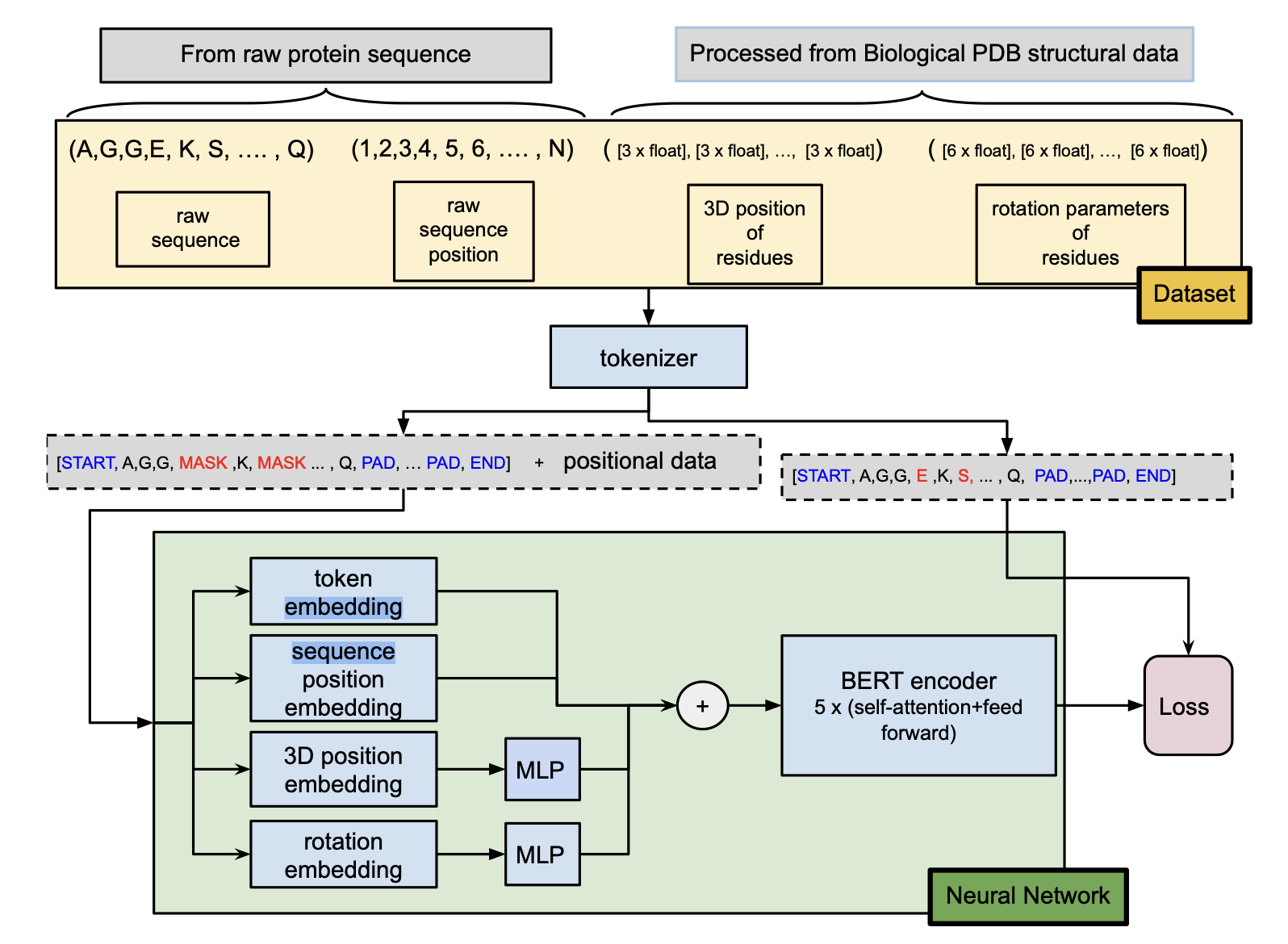
本文从NLP框架的角度将蛋白质视为句子,将残基视为单词/符号并使用轻型BERT模型对蛋白质数据进行编码。与纯粹的NLP模型的主要区别是使用额外的嵌入来为模型提供三维信息。除了原始Transformer文章中描述的经典token嵌入和位置嵌入外,本文还加入了两个额外的嵌入模块,3D位置嵌入以及旋转嵌入,随后紧接dense层。
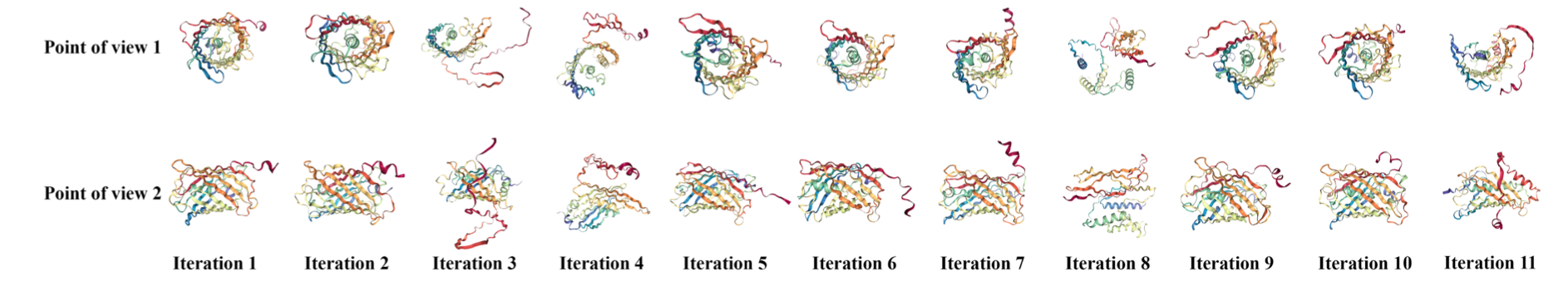
上图展示了生成过程,目标是生成一个与给定target共享相同蛋白质结构的新蛋白质。为了进行生成,本文通过检索目标蛋白、其主要序列和结构数据(已知或用Alphafold预测)来初始化。然后对序列中的每个残基(在下文中表示为n)进行循环,具体结合吉布斯采样的流程如下:
- 遮蔽主序列中当前的氨基酸n数据。
- 将主序列和结构数据送入PeTriBERT,它将返回一个预测分布p(tn|t1, ...tn-1, tn+1, ...tN , c),给定其他氨基酸p(tn|t1, ...tn-1, tn+1, ...tN ) 和结构数据c。
- 从这个分布中取样,用序列中的结果替换ti
- 这个循环重复多次,每一次都用前一次的输出进行初始化。另外,在每个循环的末尾存储所得到的候选序列。
- 当生成结束后,将所有生成的序列给到AlphaFold,并选择一个获得最高lDDT分数的序列。
具体在avGFP蛋白上,本文使用吉布斯抽样进行了2组不同的实验。
- 使用avGFP序列作为目标结构生成200个蛋白质,与上文中描述标准流程完全一致。
- 使用avGFP序列生成200个蛋白质,其中第65、66和67位的氨基酸被强制fix为原始序列残基,分别为S(丝氨酸)、Y(酪氨酸)和G(甘氨酸),这些残基对于蛋白质的荧光特性来说是至关重要的。
当算法结束时,400个生成的蛋白质序列通过AlphaFold进行处理,并计算每个候选蛋白质与参考的avGFP蛋白质之间的lDDT得分。
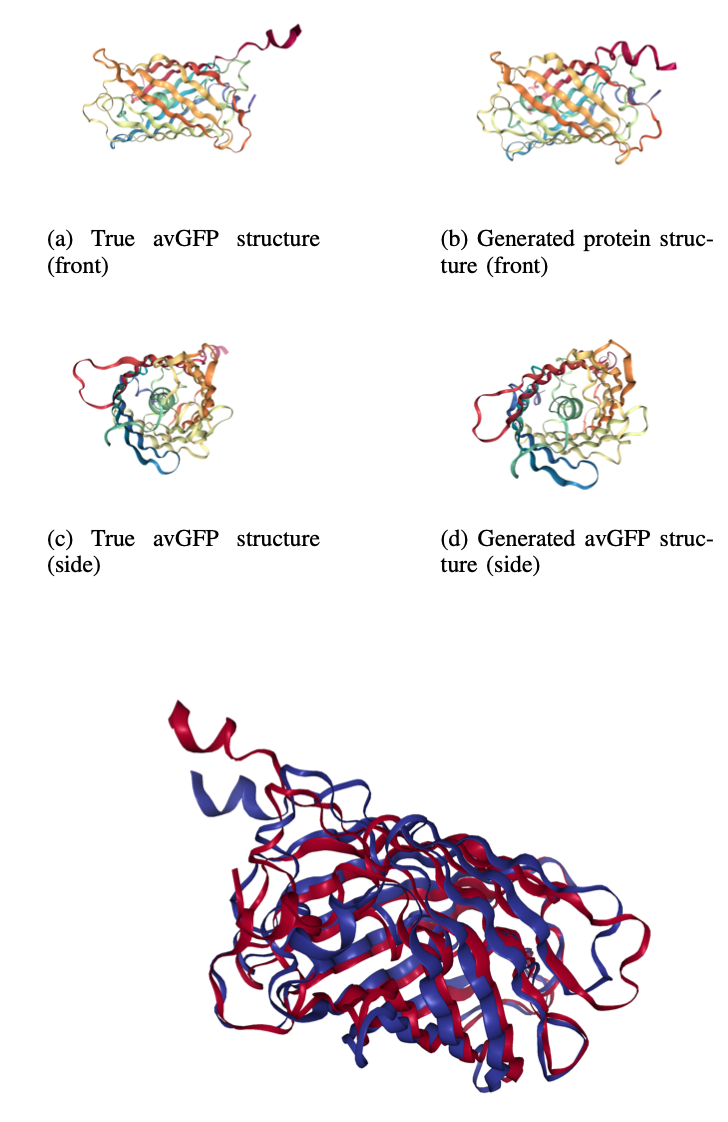
最终结果中,作者保留2组中的每组五个最佳得分的蛋白质,得到的10个蛋白质的IDDT得分范围是0.70到0.79,每个蛋白质的序列相似度约为20%。当对整个蛋白质组进行blast时,10个合成GFP中有9个没有返回同源物,这表明PetriBert能够设计出具有给定三维结构的纯合成蛋白。
创新点
- 尽管是一个小模型,但本文所提出的PeTriBERT逆折叠模型能够很好地捕捉AlphaFold数据,因此,当它与吉布斯采样结合时,不需要额外的学习就可以用于蛋白质设计。
- 本文通过生成与avGFP结构非常接近的蛋白质序列对PeTriBERT的结果进行了基准测试,并讨论了合成蛋白质的虚拟进化。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢