
【论文标题】SHINE: Protein Language Model based Pathogenicity Prediction for Inframe Insertion and Deletion Variants
【作者团队】Xiao Fan, Hongbing Pan, Alan Tian, Wendy Chung, Yufeng Shen
【发表时间】2022/09/02
【机 构】哥伦比亚大学
【论文链接】https://doi.org/10.1101/2022.08.30.505840
【代码链接】https://github.com/xf-omics/SHINE
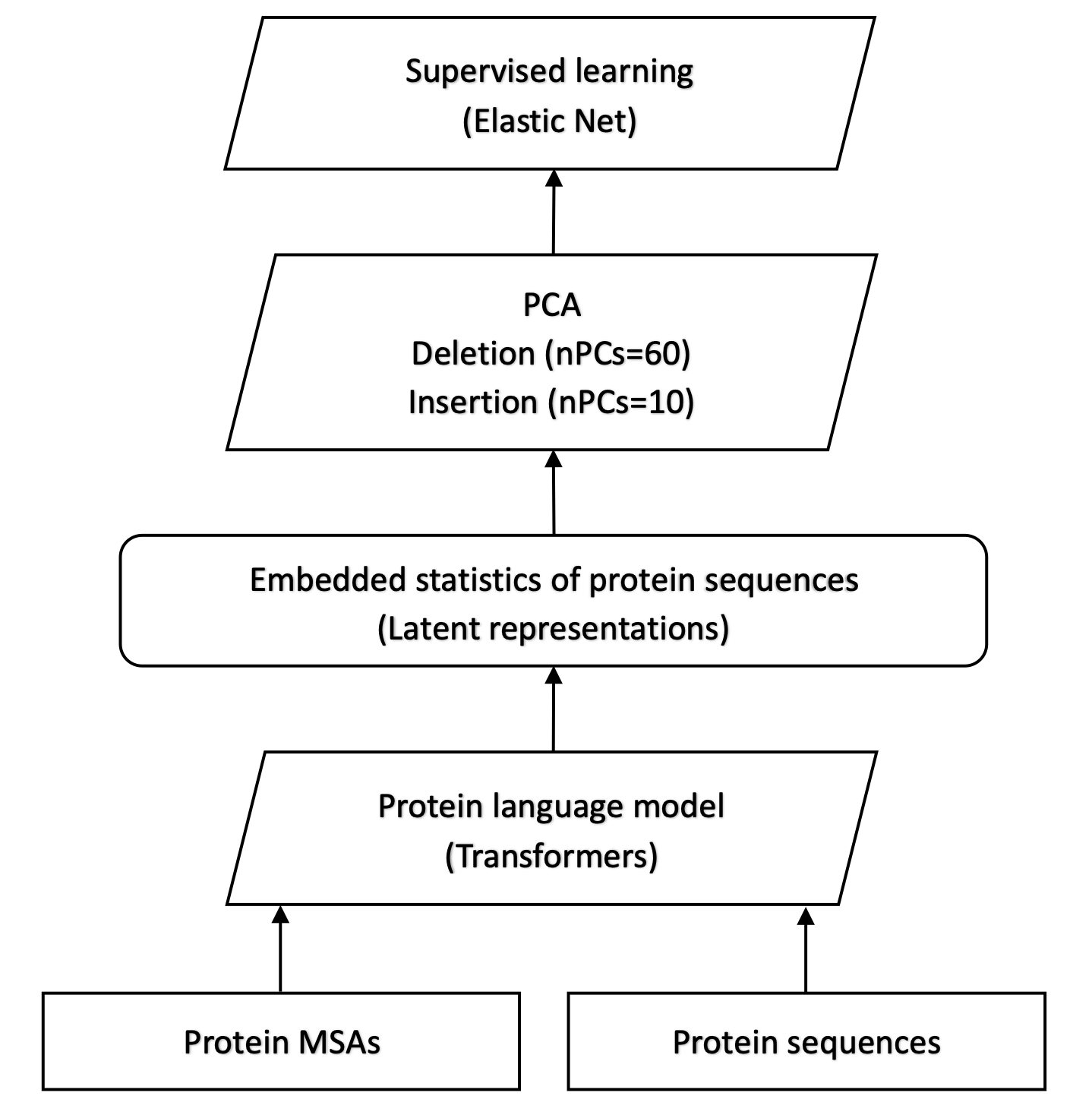
上图展示了SHINE使用的迁移学习架构,利用预训练蛋白质语言模型和有限的可用致病性标签来处理框内 indels。本文使用了两种蛋白质语言模型。ESM-1b和MSA transformer,ESM-1b transformer是在2.5亿个蛋白质序列上训练的,MSA transformer是在2600万MSA上训练的,两者都产生了包含输入蛋白质的生物属性信息的隐藏表征。
由于来自 transformer的隐藏表征是高维的和相关的,本文首先使用主成分分析(PCA)对来自ESM-1b和MSA transformer的1024和768隐藏表征进行了特征还原。使用线性回归作为基础预测器来选择剩余主成分(nPCs)的最佳数量。转化后的主成分作为突出特征被输入到一个监督机器学习模型中。本文进一步测试了不同的监督机器学习模型,包括随机森林、支持向量机、梯度提升和弹性网络。对于多氨基酸差异,本文计算每个氨基酸的预测分数,然后测试分数的最大值、平均值和总和作为最终预测分数。
ESM-1b和MSA transformer将蛋白质主序列和MSA作为输入。本文使用REST API(https://rest.ensembl.org/documentation/info/genetree)从Ensembl Compara下载MSA数据。MSA深度的中位数和平均值分别为211和320.2。本文对系统发育树进行了修剪,以去除不太相似的蛋白质,每个MSA最多包含300个蛋白质。这也加快了生成隐藏表征的预训练过程,修剪后的MSA深度的中位数和平均值分别为199和184.3。本文将野生型蛋白质序列或MSA送入预训练的 transformer,并提取被删除的氨基酸的隐藏表征。对于插入,野生型MSA被用于MSA transformer。提取的是位置(氨基酸或间隙)的潜伏隐藏表征,然后是插入发生的氨基酸。具有插入氨基酸的突变蛋白质序列被输入ESM-1b transformer,插入的氨基酸的隐藏表征被用作特征。
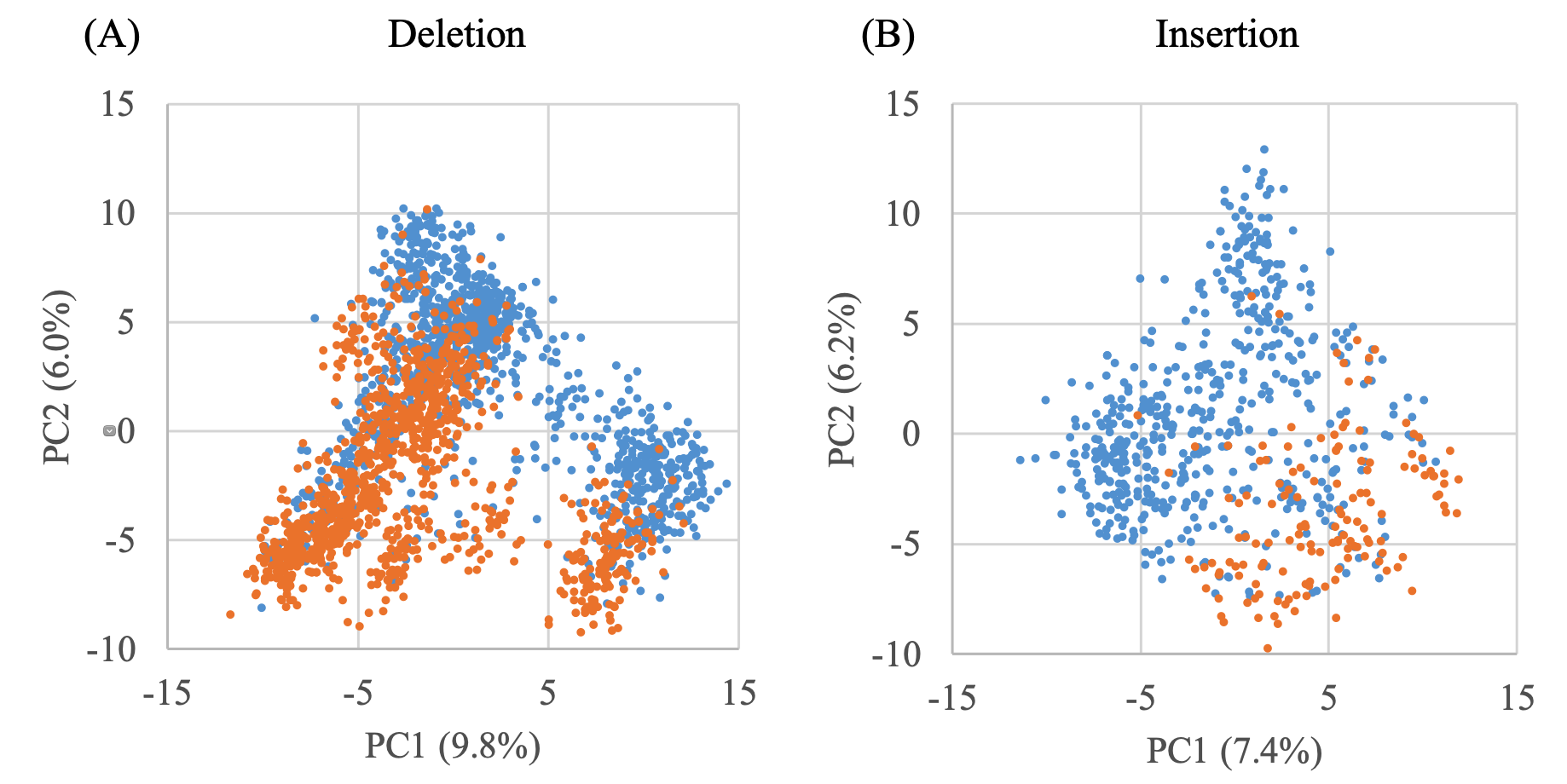
上图显示了致病性和良性突变的前两个PC(主成分)的散点图。PC1和PC2与缺失的致病性相关,相关系数为-0.376和-0.464,对于插入,PC1和PC2的相关系数分别为0.482和-0.415,前10个PC分别解释了删除和插入表现的41.0%和40.3%的突变。本文为删除和插入选择了80个和10个PC,因为它们在线性回归模型的基础上给出了最高的AUC值。
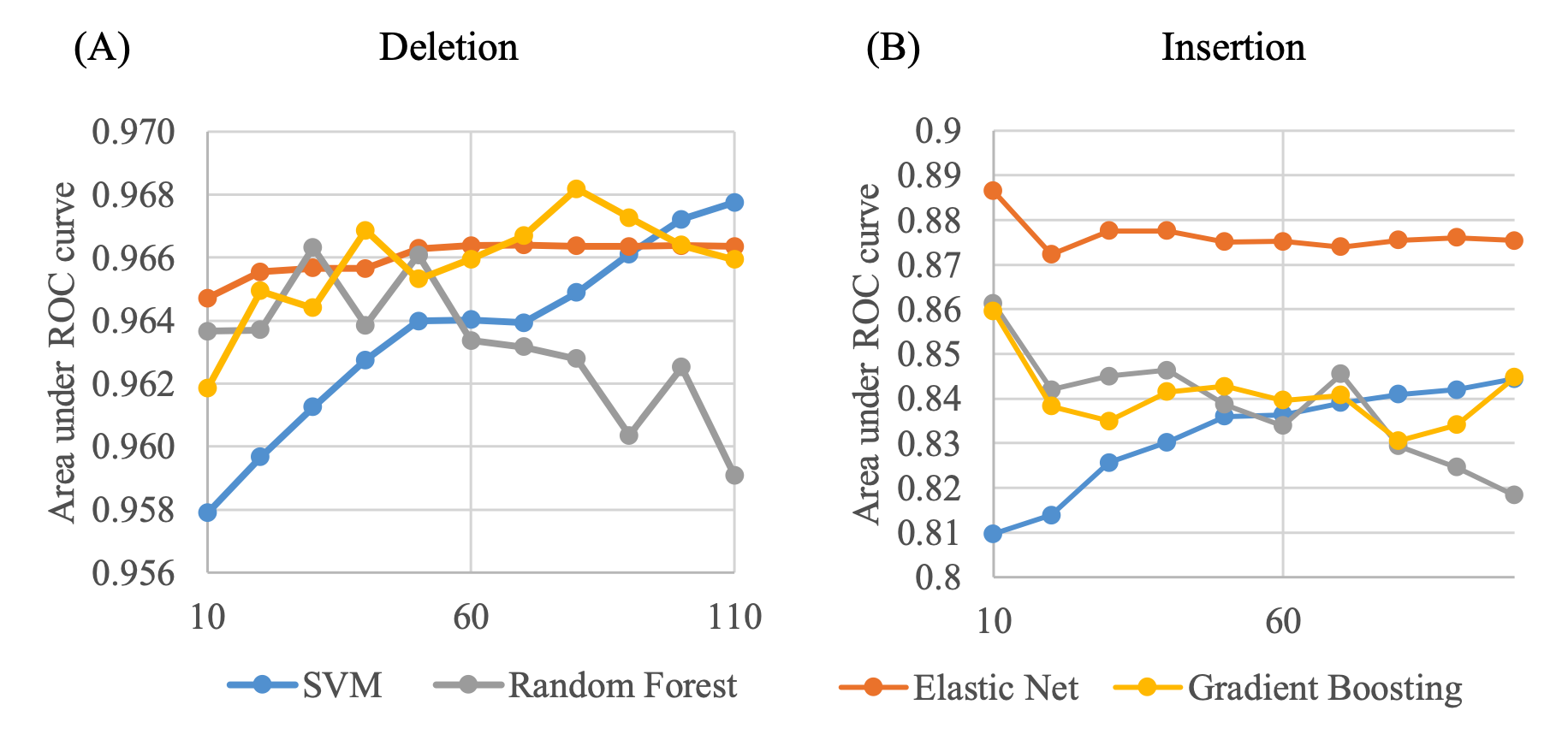
上图展示了使用不同PC作为输入的机器学习模型的AUC。本文测试了不同的监督机器学习模型,并调整了它们的参数,将最后一步优化的相同的nPCs作为输入。最后,每个调整过的模型都被用来根据最佳AUC值选择它们自己的最佳nPCs。
本文选择了弹性网络,因为它们提供了一致的良好性能,对输入PC的数量不敏感,并且不可能在训练数据集上过度拟合。弹性网络的参数α和l1_ratio对于删除和插入都是0.5和0.1,删除包括60个PC,其中24个的系数不为零,插入包括10个PC,它们都有非零的系数。
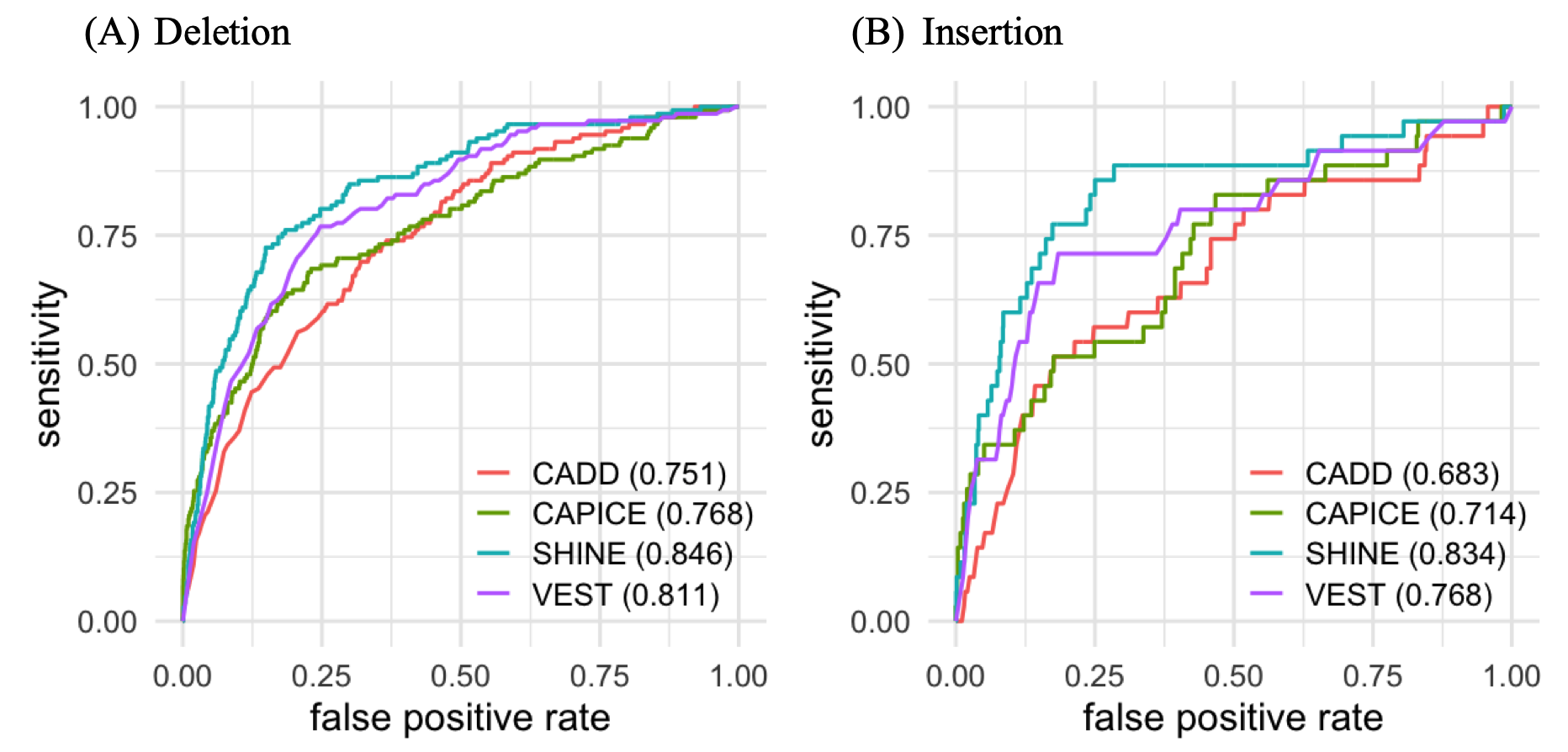
上图展示了在NDD案例上的结果。从ROC曲线来看,SHINE的AUC值最高,分别为0.846和0.834,比第二好的VEST-indel在删除和插入方面分别提高了0.04(相对提高4.4%)和0.07(相对提高8.5%)。与其他三种方法相比,这种改进是显著的(P值<0.05)。在SHINE的低假阳性率下,敏感性迅速上升。这意味着SHINE的分数与致病性的可能性相关。本文使用他们的默认阈值评估了他们的二元预测。总的来说,SHINE和VEST提供了平衡灵敏度和特异性的良好准确性。
本文的分析表明,SHINE可以很好地区分致病性和良性,对于高精度预测提供了一个很好的解决方案。
创新点
- SHINE是第一个基于蛋白质语言模型的方法,用于预测框内indels的致病性。蛋白质语言模型以无监督的方式产生无偏见的蛋白质统计数据。
- 未来的研究应该考虑使用类似的方法扩大致病性预测的突变类型。
- 随着对框内 indels的突变扫描数据对框内 indels的出现,来自功能数据的基准数据集将受到高度重视。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢