
近日,索邦大学、巴黎大学、巴黎城市大学巴斯德研究所、以色列特拉维夫大学联合发表了题为“Computational protein design with evolutionary-based and physics-inspired modeling:current and future synergies“的蛋白设计综述文章。
蛋白质设计的机器学习方法正沿着两条平行轨道快速发展:基于进化的方法和物理启发的方法。两种方法具有高度互补性。
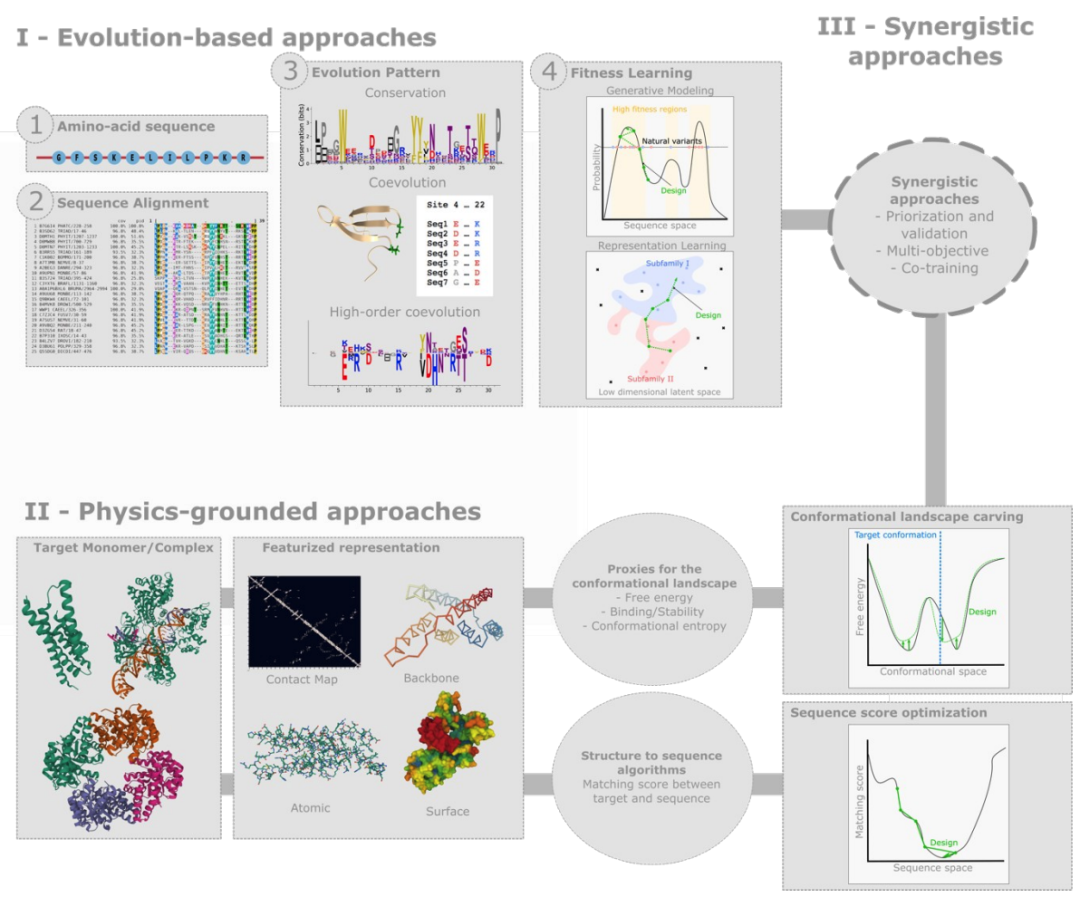
基于进化的方法,前者推断具有所需结构或功能的蛋白质序列集合共享的序列特征。后者使用机器学习surrogates估计关键生化特性,例如结构自由能、构象熵或结合亲和力。
总结
1 基于进化的方法
(1) 目标特性的显著优化需要探索含有来自野生型蛋白的许多突变的序列。然而,据估计多达50%的单点突变对功能有害,导致多个位点突变时成功率呈指数下降。
一种解决方案是将搜索限制在蛋白质自然进化过程中先前遇到或可能遇到的突变或突变组合。例如,Russ等人[9] 利用DCA设计了数百种不同的具有天然功能的脊索酸变位酶,成功率高(~30%)。最近各种机器学习生成模型,在各种酶和纳米设计任务中取得了成功[10,11,12,13,14]。
(2) family-level模型缺点:不能在跨蛋白质家族中得到泛化,因此需要大量序列进行训练。
克服这些限制的一个可能途径是蛋白质语言模型,因为它们可以同时模拟不相关的蛋白质序列集。
2 物理启发的方法
(1)蛋白质设计问题相当于在规定的构象状态下(作为单体结构,与配体结合)找到具有低自由能的序列。基于力场的蛋白质设计的局限性包括计算成本高(基于蒙特卡洛的优化速度慢且效率低)、骨架结构的序列恢复率不令人满意(30%-50%)以及实验成功率有限。
- a) Norn等人提出将负对数似然解释为自由能的代理。他们用模型预测的几何特征的概率来近似玻尔兹曼系综中构象的概率。对于具有多个低能构象的序列,trRosetta预测的几何特征分布通常很宽或多峰。因此,trRosetta估计的自由能比Rosetta能量更好地预测折叠到目标构象。
- b)模型预测分布的香农熵可以作为构象玻尔兹曼分布物理熵的代理。
(2) 基于物理的方法的两个基本挑战:1)需要对结构构象空间进行广泛采样以估计热力学量,以及2)探索巨大序列空间的高计算成本。
基于AlphaFold2, AlphaFold2-multimer和RoseTTAfold的幻觉方案其中预测不确定性(pLDDT或PAE)可以用作构象熵的代理。
3 基于进化和物理启发的协同建模
(1)进化模型可以用于快速生成不同的序列库,然后,根据计算密集型物理启发模型中获得的分数对候选序列进行优先排序。
(2) 训练MSA和结构的联合模型。使用进行信息对基于结构的序列生成模型进行微调,或者相反,使用结构信息对进化模型进行正则化。
(3) 将已知结构信息合并为先验,例如使用结构感知transformer模型,如EvoFormer(其中结构作为模板提供)。
(4) 基于ML的分子动力学[60]或神经力场[76]的未来发展可能为理解目标蛋白的高亲和力结合物的合理设计中的挑战,扩展到其他类型的配体提供了重要动力。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢