
AI蛋白质结构预测赛道,国产模型又有吸睛表现:
在蛋白质结构预测竞赛CAMEO上,有支队伍连续四周夺得全球第一。
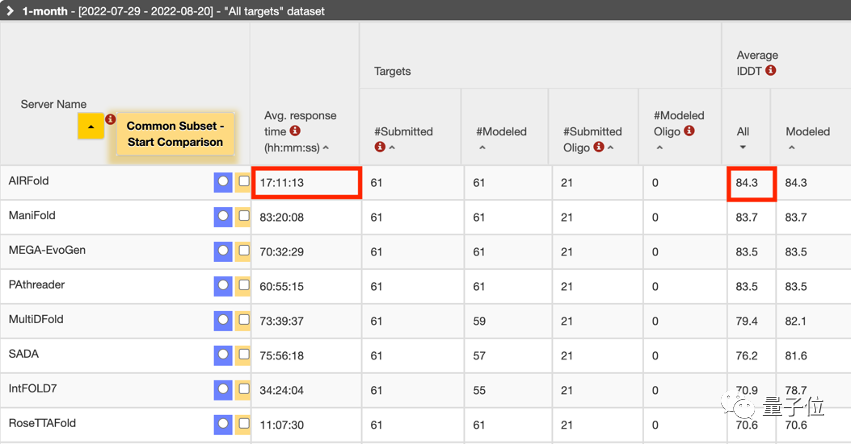
达成这一成就的是来自清华大学智能产业研究院(AIR)的AIRFold。
△AIRFold 在7.23-8.20的评估中连续四周全球第一
CAMEO竞赛(Continous Automated Model Evaluation)与CASP并列为蛋白质结构预测领域的两大权威竞赛。
不同之处在于CASP两年一届,CAMEO则是持续举办,每周都有结构生物学家最新破解出的蛋白质结构作为赛题。
CAMEO上得分与排名每周实时更新,华盛顿大学David Baker团队的RoseTTAFold、百度腾讯华为等行业顶尖选手都在其中参与角逐。
AIRFold在近4周的比赛中,不仅预测结果IDDT分数领先,系统响应时间上也远远领先后几名的团队。
亮眼成绩如何取得?后续又有哪些研究和应用方向?
带着这些问题,我们联系到项目负责人清华大学智能产业研究院清华大学智能产业研究院(AIR)的兰艳艳教授,与她进行了深入交流。
下面送上对话实录,为方便阅读,我们在不改变原意的基础上做了编辑整理。
对话实录
量子位:AIRFold项目是从什么时候开始做的?能否介绍一下团队基本情况?
兰艳艳教授:AIRFold项目是AIR智慧医疗方向的一个重要部分,大约是2021年9月份开始,距离现在刚好一年左右的时间。
团队成员是陆续到位的,目前总共有7-8人,除我之外还包括科研工程师,博士后和博士生等。成员背景基本都是AI方向的,也有生物学和化学背景的同学参与。
张亚勤老师和马维英老师在整个项目进行过程中也一直在帮我们把握方向,提供资源支持,给我们团队很多指导。同时我们的访问教授彭健老师以及他带领的Helixon(华深智药)团队也和我们进行过多次讨论,对我们进行了一些技术指导,帮助我们团队得到了很好的成长。
量子位:在AlphaFold2之后,出现了单序列预测的一些方法,AIRFold为什么坚持走同源信息挖掘这条路线?
兰艳艳教授:OmegaFold和ESMFold等单序列模型确实没有显式地使用MSA作为特征,但严格讲并非没有用同源序列中隐含的共进化信息,它采取了一种隐式的使用方式。具体来说,OmegaFold通过基于掩码语言模型(Mask Language Model, MLM)的蛋白质预训练模型编码了主序列然后用于预测结构,MLM天然地具有捕捉共进化信息的能力,这点在早期Meta 的蛋白质预训练工作ESM中也有体现。直接使用MSA或者使用具备捕捉共进化能力的编码器都是不同的方法而已。
我们选择同源挖掘路线主要有几个原因:
第一,从效果上讲,以Meta(原FAIR)的ESMFold为代表的基于单序列的结构预测方案,比基于单序列的AlphaFold2效果要好,但是与直接显式使用MSA序列的AlphaFold2方法相比还有不少差距。例如ESMFold在CAMEO以及CASP数据集上测试所得的TM-score分别是82.8以及67.8,对应AlphaFold2的TM-score是88.3以及84.7,有较大差距。我们认为ESMFold确实给我们指明了利用同源信息的新方式,但要达到替代MSA的效果还有较大的改进空间。
第二,当时选择同源挖掘这条路线,首先是因为我们团队有丰富的NLP背景,我们一看到AlphaFold2,就发现MSA这个模块作为同源信息的输入非常关键,而AlphaFold2的使用方式还停留在传统方法上,因此我们很自然会优先选择从我们擅长的MSA序列建模和检索这个方面入手,应用最新的NLP技术来进行突破。
第三,最重要的原因,我们做AIRFold和参加比赛最终的目的不仅仅是为了蛋白质结构预测本身。我们希望在这个过程中从建模和计算的角度探究哪些重要信息如何作用最后影响了折叠的结果,这些积累能够锻炼我们的队伍,让大家对结构预测这个问题有更深刻的认识,同时也会启发我们对蛋白质相互作用、序列到结构到功能等问题的思考,从而促进我们开展与AI赋能新药研发相关领域的研究和应用。
量子位:能否展开讲讲同源挖掘模块Homology Miner的技术细节和特色?
兰艳艳教授:挖掘同源信息是目前主流蛋白质结构预测模型以及参赛服务器都会关注的一个关键技术方向,AIRFold的特色集中在获取同源蛋白和对同源蛋白进行优化校正的方法上。
AIRFold 的Homology Miner在经典的同源检索算法之外,整合了一些基于NLP全新技术所形成的算法,包括稠密检索、面向多序列比对的同源蛋白生成等模型,这一系列的方法在一些初始缺乏同源信息的孤儿蛋白上,展示了比较明显的效果,说明目前主流的同源检索方法存在可提升空间。
除此之外,我们针对“什么是好的同源蛋白”这一问题,从信息论的角度给出了一个量化的定义,基于这一量化指标对于同源表征进行优化,可以稳定地提高结果以及鲁棒性,这也为同源表征学习也提供了一个全新的思路和角度。
量子位:除了同源挖掘模块外,AIRFold在AlphaFold2的基础上还做了哪些改进?
兰艳艳教授:除了同源挖掘模块,我们对AlphaFold2目前所存在的一些问题也进行了有针对性的探索和改进。
例如在结果预测上,AlphaFold2还无法很好的解决蛋白的多构象和点突变问题,模型精度(即pLDDT)的预测也存在偏差的问题等。
以pLDDT的偏差为例,pLDDT本身是结构预测结果的一个置信度,大家发现在AlphaFold2提供的预测结果中,pLDDT通常还是比较准的,高的地方预测结果相对比较准确,低的地方预测结果不够好,但是事实上作为神经网络的输出结果,pLDDT的鲁棒性很差,很难反映MSA的微小变化或攻击带来的影响,这样就导致在比赛或者实际应用中,如果完全以pLDDT为标准进行选择,会引发严重的问题。
在这方面我们也提出了一些对抗训练,多目标优化等新的思路,在这些问题的解决上取得了一些进展,欢迎大家关注我们后续的科研工作。
量子位:AIRFold保持高IDDT评估的同时,在系统响应时间上远远领先其他团队,是靠什么做到的?
兰艳艳教授:AIRFold是一个自动化的平台,包括同源序列增广、同源序列筛选、特征处理、结构预测、结果分析以及自动提交等模块。
比赛序列过来的时候没有任何人为的干预,我们的系统会自动的监控server是否有新来的序列,自动的补上提前设置好的参数配置,自动对蛋白质结构进行预测最后提交预测结果。
我们设计并实现AIRFold的初衷就是为蛋白质结构预测以及同源蛋白分析这一问题,找到通用的解决方案。尽管在比赛中的序列之间差异很大,比如有的同源很多,有的同源蛋白很少,我们在比赛中始终坚持使用同一套策略和系统,来减少人工对于不同的比赛序列进行不同的处理,力求得到一套通用的结构预测解决方案,这是我们响应迅速的主要原因。
量子位:AIRFold团队重点介绍了CAMEO比赛中一个较难预测的蛋白7TVI,它的预测难点在哪里?
兰艳艳教授:7TVI是来自 Planctomycetes(浮游菌门)细菌的Cas13bt3蛋白,相比其他序列来说有两个难点。
首先是这个序列同源序列相对少,多序列比对(Multiple Sequence Alignment ,MSA)结果无论是从深度还是覆盖度来说都是非常不理想的。我们第一次搜索的结果只搜索到了700条左右的同源序列,有三分之二以上的序列基本没有覆盖。我们都知道当下流行的AlphaFold2是高度依赖同源信息的,这样低质量的同源序列必然是不利于结构预测的。
AIRFold团队充分利用自主研发的HomoMiner的优势,对低质量的MSA进行筛选过滤,选择其中有价值的部分,去除冗余;同时利用深度稠密检索技术和同源序列生成技术对MSA进行补充,丰富其中的信息,因此能在这个序列上做出比较好的结果。
其次,这个蛋白结构域多,变构大,因此构象比较复杂灵活。从PDB的结构和以往的研究来看,这个蛋白有HEPN1和HEPN2两个核酸酶结构域,crRNA结合结构域又分为Helical1-1,Lid,Helical1-2,Helical2和Helcal1-3五个,中间有linker连接。目前主流的结构预测方法,比如AlphaFold2和ESMFold都主要使用单结构域蛋白进行训练,这是因为PDB数据库中单结构域蛋白远多于多结构域蛋白。
此外,MSA中往往也会出现每条序列只覆盖一个结构域的情况,不能提供多结构域之间关系的信息。这就造成多结构域之间关系不容易被准确预测。
AlphaFold-Multimer的成果对我们很有启发,秉承结构域间关系信息也蕴藏于MSA中的理念,我们使用HomoMiner对MSA进行筛选,去除信息量低、噪声大的序列,提高高质量同源序列中长程相互作用信息的信噪比。因此我们可以更准确地建模多结构域之间的关系。
这些经验也符合我前面说到的,充分挖掘MSA信息虽然更加耗时,但是在实际数据上能提供更具启发性的结果,也能够让我们在此过程中有思路去分析蛋白质的进化生物学问题。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢