
近日MIT的Matthew E. Walsh、Tristan Bepler等发表预印本论文展示了一种端到端的基于贝叶斯优化及语言模型的方法来设计大型多样化的抗体单链可变区(scFv)库。在与传统定向进化方法的正面比较中,本文的方法产生的最佳scFv比定向进化的最佳scFv的亲和力提高了28.8倍,此外,在最成功的文库设计中,99%的设计scFv都比初始scFv有改进。本文方法并不假定候选scFv与靶标强烈结合,并且依赖于序列数据而不需要序列比对或靶标抗原结构,同时还可以结合疏水性、等电点等抗体性质预测方法,适用于任何靶标抗原的早期阶段的抗体开发。
1.
背景
在新冠流行的当下,针对性抗体研发十分重要,如何设计高质量的抗体库是抗体研发中备受关注的问题,其难点在于如何有效的进行针对巨大抗体空间的探索。目前应用广泛的基于免疫方法筛选出的抗体库往往只占整个抗体空间的极小部分,且往往在亲和力或可开发性上存在问题,需要再次优化,而优化过程往往受结构解析等实验的成本与效率的问题。
AI技术近几年在上述问题上崭露头角,通过alphafold等结构预测方法或esm等蛋白表征方法在药物发现和设计上展现了巨大的前景,在抗体方面,igfold,antiberta等类似的AI工具也可供使用。在抗体库生成设计方面,现有基于深度生成模型的方法,虽然这些方法可以产生全新序列,但无法不能针对特定靶点进行生成,并且也无法对库的质量进行评估并进行序列优化,本文的方法即针对以上问题提出了有效的解决方案。
2. 方法 .
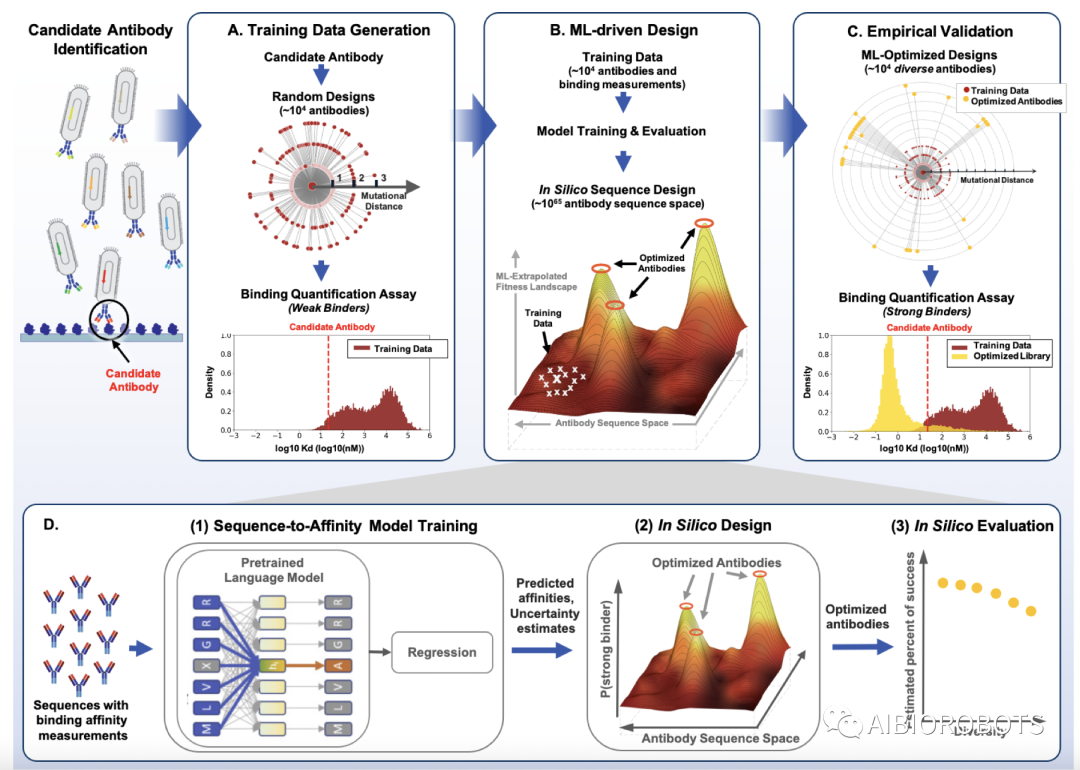
本文开发了一个端到端AI驱动的scFv设计框架,它结合了预训练语言模型、贝叶斯优化和高通量实验,可以直接设计整个scFv链。本文方法优点在于只需序列数据,不需要序列比对或对靶标抗原结构的了解,从而适用于针对任何靶标抗原的抗体开发的早期阶段。
具体方法分5步
1
对候选scFv的CDR区域进行随机1-3位点突变,并进行酵母交配实验高通量测定以获得监督的训练数据,包括17,118个重链scFv亲和力数值和26,223个轻链scFv亲和力数值。
2
在大量的蛋白质序列上进行语言模型的无监督式预训练,以提取scFv表征,本文总共使用了4个预训练模型,分别为五千万Pfam数据上的蛋白质语言模型、和OAS上三亿抗体重链预训练的模型、七千万抗体轻链训练的模型和三万配对的轻重链数据训练的模型。每一个数据集上的模型架构都选择24层的BERT进行序列的遮蔽预测,以困惑度作为评价指标之一。
3
在实验测得的四万带标签训练数据上对预训练的语言模型进行监督式微调,以预测结合亲和力,在不确定性度量上本文使用了集成或高斯过程(GP)。这里的集成模型由16个不同的回归模型组成,通过4种不同语言模型加上2种不同的损失函数(平均平方误差MSA和平均绝对误差MAE)以及这两种不同的数据预处理步骤(实验重复三遍,其中缺失值放弃或者用其他数据中位数填充)所进行组合得到的。对于GP模型,本文将预训练语言模型中每个氨基酸的向量表征连接起来,然后进行主成分分析将向量维度降维到1,024维并在其上训练GP模型。
4
从训练好的序列-亲和力模型中构建基于贝叶斯的scFv适应度景观,然后通过贝叶斯优化验证进行scFv设计,采样算法采用了爬坡(HC)、遗传算法(GA)和吉布斯采样。与序列直接映射到亲和力的非贝叶斯方法不同,本文适应度函数被定义为从整个scFv序列到后验概率的映射,后验概率定义为预测结合亲和力优于训练集平均亲和力的概率,并可从集成模型和GP模型中获得。不同抽样算法的处理方式如下:
+ + + + + + + + + + +
爬坡
从亲和力数据集中的最强的10条序列初始化,随机单双位点突变优化,每次迭代根据局部搜索结果根据适应度进行贪婪采样直到到达局部最优值,这里局部搜索空间定义为当前序列的1000个突变体,由所有单突和随机双突组成。
+ + + + + + + + + + +
遗传
同样从亲和力数据集中的最强的10条序列初始化。根据Wright-Fisher进化模型,从当前种群中选择父母,当前种群成员成为父母的概率与他们的适应度成指数正相关,随后对从亲本种群中随机选择的两个亲本序列进行随机单突组合获得子本序列,重复直到不再产生新的序列。
+ + + + + + + + + + +
吉布斯
有所不同,从亲和力最强序列进行初始化。每一步随机选择序列中的一个位置,以其他氨基酸为条件计算的适应度条件概率对所选位置进行采样,并通过用采样的氨基酸替换原氨基酸以更新序列,总共迭代30,000次。
05
实验验证各个库中预测具有强亲和力的前6000条scFv序列。对比的则是定向进化基线模型选择随机突变库和PSSM采样库。
3结果
●3.1 抗体库的亲和力水平和多样性
上图展示了每个库在轻重链设计上的生成结果,包括最优亲和力数值、突变数以及相比较于传统方法PSSM生成的库的提升幅度。可以看到来自机器学习优化库的最佳scFv明显比来自PSSM库的scFv有更强的结合力,而且一般有更多的突变。结合力最强的重链设计来自En-Gen库,比PSSM库中最强的scFv结合力强28.8倍。最好的轻链设计是在En-Gibbs库中,比PSSM库中最好的scFv高了7.8倍。
对预测模型同样重要的是采样算法的选择,算法对探索性能和多样性之间的权衡很重要。当使用集成模型的适应度景观来设计14-Ab-H时,爬坡和遗传算法发现scFvs的结合力比最佳PSSM采样的scFv有明显的增加,分别为28.8和28.2倍,而且这两种方法的总体成功率都非常高。然而吉布斯取样算法无论采样的最佳亲和力还是总体成功率都稍逊稍逊一筹,但是该方法生成多样性却特别高,展示了多样性与最优亲和力之间存在的平衡。但是当设计轻链时,En-Gibbs组合得到了最强的亲和力,多样性方面也有23个突变,这展示了多样性探索的收益。
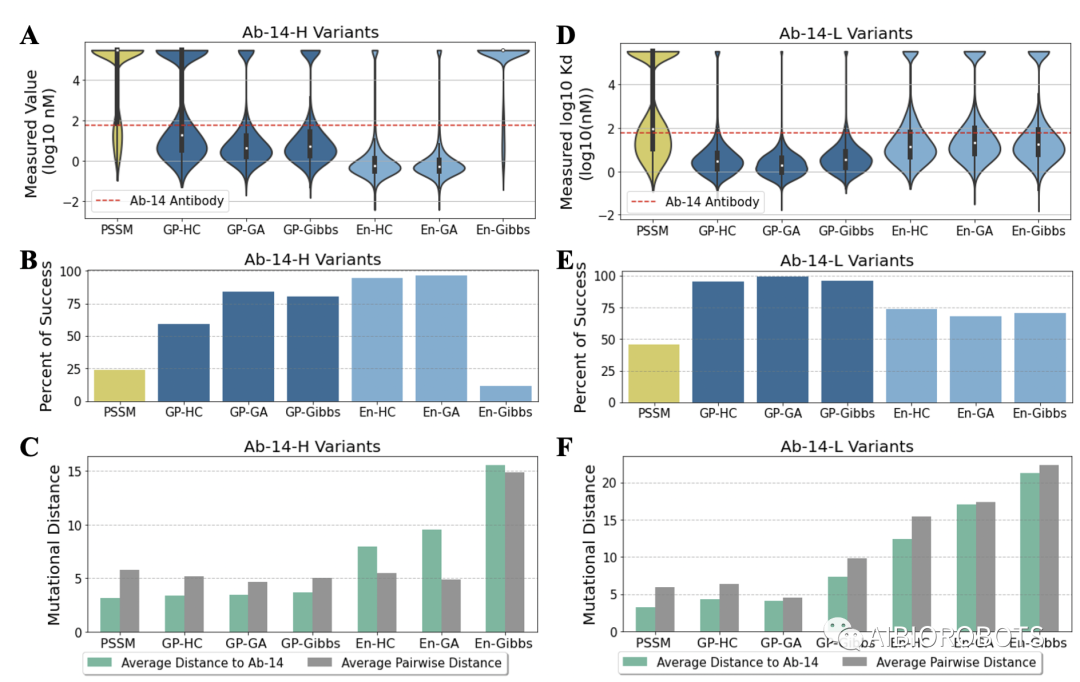
多样性评价上的评价指标为两个,与初始序列的距离以及衡量数据内部序列间的平均成对距离。总体上机器学习优化的库多样性好与PSSM库。对于Ab-14-H重链设计,集成方法的效果最好,尤其是En-Gibbs库,但是这个库在亲和力上的表现很不好,说明存在多样性和亲和力优化之间的探索/利用的平衡,轻链设计结论类似。
上图显示了各个设计库的性能和多样性,其中性能评价指标为亲和力分布和成功率,本文的成功率定义为生成scFV比初始scFV具有更高亲和力的比例。对于Ab-14-H重链设计,除了En-Gibbs库中的序列外,所有机器学习优化的库在结合亲和力中位数上和成功率都超过了PSSM库,其中En-HC库成功率94.3%,En-GA库成功率96%,效果较好。对于Ab-14-L轻链设计,与PSSM库的比较上结论类似,不过基于GP的库的成功率表现是最好的。
3.2 理化性质的多样性
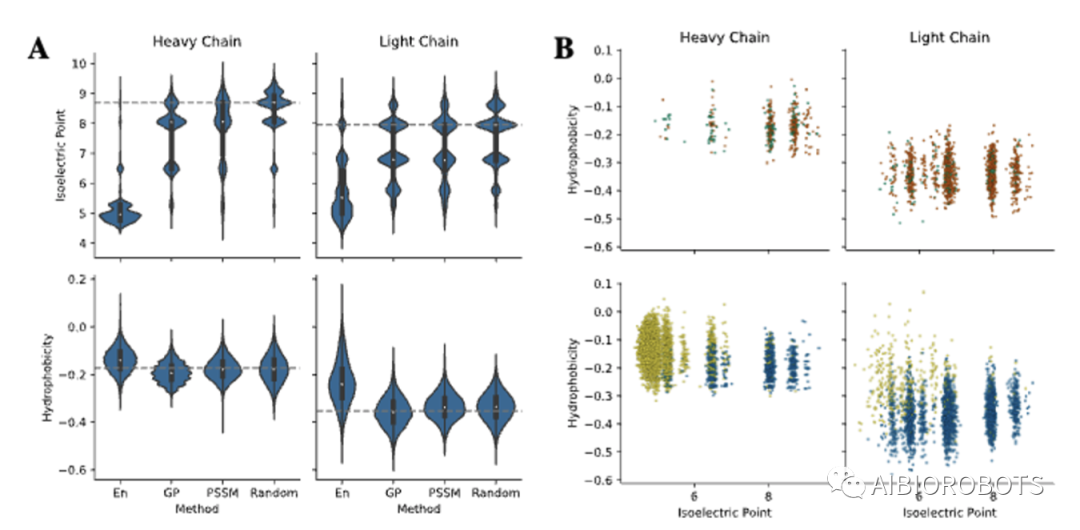
本文使用BioPython根据每个库中序列计算生物物理性质,左图展示了等电点、疏水性在突变库、PSSM库、集成库和GP库当中的分布,右图展示了实验亲和力小与1nM的序列的生物物理性质的联合分布,其中上侧的随机(橙)库、PSSM(绿)库具有和下侧的集成方法(黄色)、GP(蓝)相似的分布。结果表明本文方法生成了具有多样性理化性质的序列库。
3.3 突变距离的分析

上图展示了两类序列-亲和力模型,集成模型和GP的性能,比较了模型预测和实验值的Spearman相关系数和MAE平均绝对误差。可以观察到,集成模型在预测亲和力方面比GP模型做得更好。当对测试集进行评估时,重链和轻链集成模型的Spearman相关系数都比GP模型略高,当对设计的Ab-14-L突变体进行评估时,轻链集合模型也略胜一筹,Spearman相关度为0.61。最明显的区别是在对设计的Ab-14-H变体进行评估时,重链集合模型的Spearman相关度为0.70,而重链GP模型的表现明显较差,这可能是由于GP模型对分布外数据的预测能力不足。
本文也评估了预测模型在设计序列上的有效突变距离,重链GP模型在离Ab-14-H有6个或更多突变的序列上的MAE急剧增加,而轻链GP模型在离Ab-14-L有10个或更多突变的序列上的MAE急剧增加。集成模型随着突变距离的增加,MAE没有明显的增加,这表明集成方法比GP模型更适合用于高阶突变体。
3.4 分布可视化
上图展示了随机突变库,PSSM采样库以及En-Gen库生成轻重链序列的亲和力分布,可以看到在轻重链上均显示,本文方法对于高亲和力抗体的富集作用,其效果具有超越传统定向进化方法的潜力。

通过tsne降维后进行可视化生成库的分布后,可以观察到以下几点:
【1】PSSM库最接近训练数据,而基于集成的库离训练数据最远。
【2】机器学习优化库位于与训练数据和PSSM库不同的子空间,说明优化算法使模型具备了外推能力。
【3】集成库的序列分布很多样化,最好的重链和轻链设计都是通过集成方法发现的,说明了基于优化算法进行探索的价值。
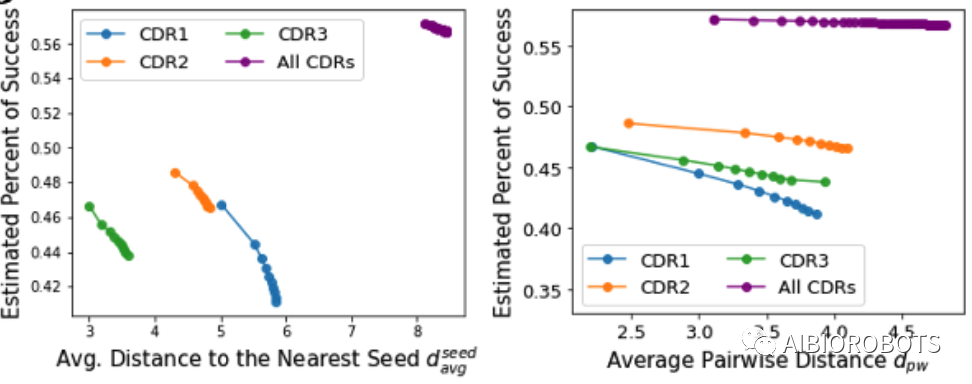
本文也比较了CDR是分别优化好还是同时优化好,通过上图可以看到,与分别设计单个CDR相比,同时设计所有重链CDR可以获得更高的成功率和多样性。
评论
本实验在机器学习方法与传统的定向进化策略的正面比较中,证明了机器学习方法所具备的优势和潜力。机器学习方法设计的scFvs具有更强的亲和力以及多样性,且在高阶突变体上亲和力预测更准。稳重的整套框架可适用于任何优化scFv性质的任务,如最小化脱靶结合或最大化中和作用等。本文通过从实验中获取高亲和力序列后利用这些数据进行监督学习来设计序列,这个思路所需的实验条件与成本较为苛刻,在方法上也有可优化空间:
1、可通过增加迭代次数而减少每轮的实验量做到节约成本
2、在初始化序列中引入生成模型也可以提高成功率
3、表位信息也可作为初始库选择的依据
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢