
【论文标题】So ManyFolds, So Little Time: Efficient Protein Structure Prediction With 预训练模型s and MSAs
【作者团队】Thomas D. Barrett, Amelia Villegas-Morcillo, Louis Robinson, Benoit Gaujac, David Adméte, Elia Saquand, Karim Beguir, Arthur Flajolet
【发表时间】2022/10/18
【机 构】InstaDeep、UCL、格拉纳达大学
【论文链接】https://doi.org/10.1101/2022.10.15.511553
近年来,用于蛋白质结构预测的深度学习方法取得了重大进展,AlphaFold有时可以接近实验精度,预示着快速蛋白质建模和设计的可能性。然而,这种模型的训练和推断需要大量的计算,而且它们非常依赖多序列比对(MSA)中包含的进化信息,而这些信息对于某些靶点来说可能并不适用。本文介绍了一个精简的AlphaFold架构,它仍然提供了良好的性能并大大降低了计算负担。与最近的方法如OmegaFold和ESMFold相一致,本文的模型最初被训练为仅从序列预测结构,利用预训练的ESM-2蛋白质语言模型的嵌入。然而将这种方法与根据MSA信息训练的同等模型进行比较,发现后者仍能提供性能提升,这表明即使是最先进的预训练模型也不能轻易取代同源序列的进化信息。本文最后训练了一个可以从预训练模型和MSA的输入中组合进行预测,或者只选择一个进行预测,并在这三种输入模式中的任何一种都获得了类似于在该环境下单独训练的模型的准确度,同时也证明了这些模式是互补的。
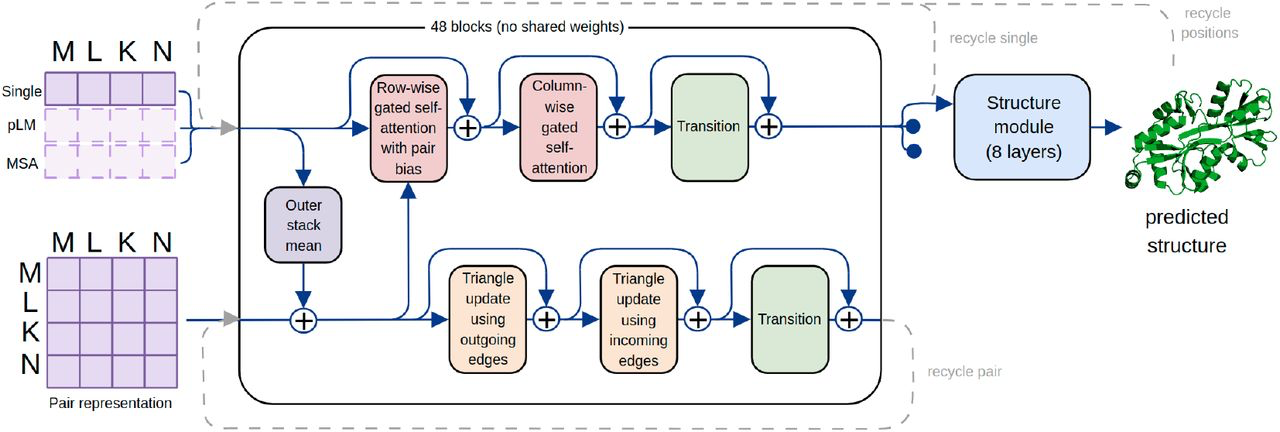
上图展示了polyfold的模型架构。该模型于Alphafold相比,完全删除了模板和额外MSA。输入信息被修改为包括一个(MonoFold)或两个(PolyFold)的预训练和MSA。除了减少一维处理轨道以匹配这些修改后的输入外,Evoformer还通过删除或精简某些昂贵的操作来减少计算负担。
输入特征方面包括:
1.预训练模型嵌入
使用650M参数的ESM-2,与ESMFold类似,对输出的每层嵌入进行投影,以获得我们的single representation。Pair representation是用成对相对位置编码初始化,再加上ESM-2的每个隐藏层的注意力图的投影。
2.MSA
为了保持与预训练模型情况下相同输入维度的轻量级架构,本文选择不堆叠来自多个MSA聚类的信息。相反采用了AlphaFold的中"无MSA "消融实验启发的方法。具体来说,single representation相当于将MSA簇的数量设置为1,簇中心是目标序列。除了成对的相对位置编码外,Pair representation是通过投影1024个原始的 "额外MSA "特征来初始化的紧接OuterProductMean模块构建的。
3.结合预训练模型和MSA输入
PolyFold将上面详述的预训练模型嵌入和MSA结合到一个单一的输入中。single representation通过将onehot编码目标序列的投影(作为第一行)与一维预训练模型和MSA输入堆叠来初始化,Pair representation是通过将预训练模型和MSA的二维初始化与成对的相对位置编码相结合而初始化的。
关于Evoformer的变化包括:
1.从一维轨道到二维轨道的通信被移到块的开头以减少训练的内存需求,并且使用OuterStackMean取代外积操作。
2.去掉了两个 "三角自注意力 "模块,只保留了 "三角更新 "模块以减少计算量。
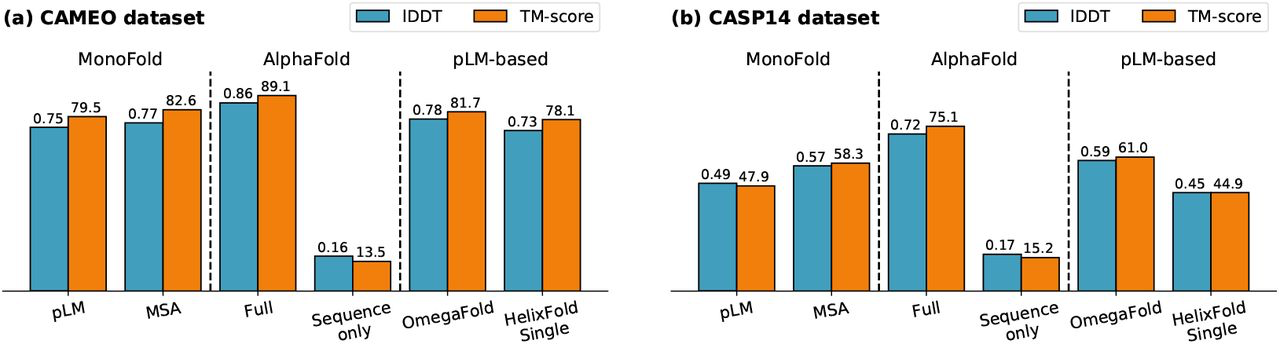
上图依次显示了monofold和polyfold的在cameo和casp14数据集上的效果。
可以发现,虽然模型进行了很多简化处理,但这些压缩模型仍然具有相当强劲的性能,MonoFold-预训练模型模型的表现优于HelixFold-Single,并且已经接近OmegaFold的性能。MonoFold-MSA甚至更好,已经匹配了最强的预训练模型模型。这表明,即使使用最好的ESM-2模型,MSA似乎仍然信息量更高,因此预训练模型还不能完全替代同源序列的进化信息。
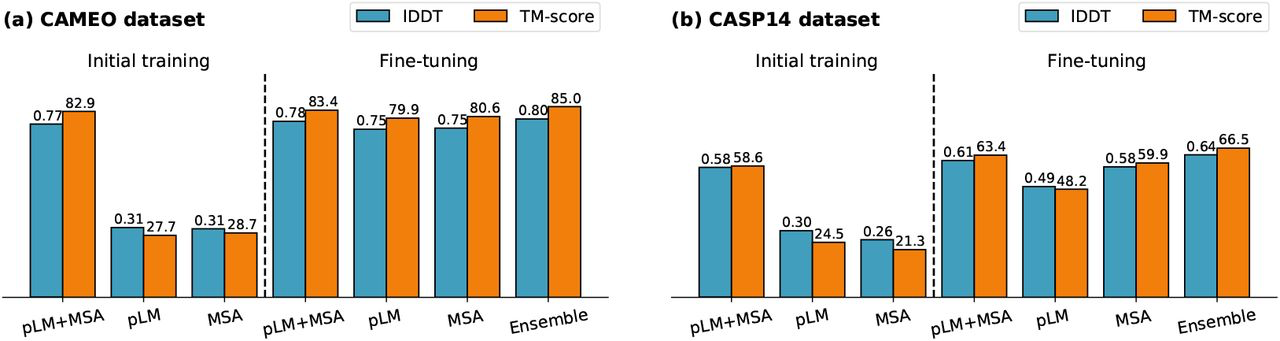
PolyFold的性能与MonoFold-MSA基本相同。这表明,预训练模型和MSA中的信息加起来并不比MSA本身更有信息性。然而,当任何一个输入被删除时,性能会显著下降,因此预训练模型嵌入不会被简单地忽略。本文也对模型进行了微调,可以看到预训练模型+MSA的准确性仅略有提高,然而,仅使用MSA或预训练模型输入的推断性能要高得多,甚至接近或达到仅在这些设置中专门训练的MonoFold模型的性能。
针对这点,有趣的是这些遮蔽信息推理(比如单MSA)经常优于全信息推理,在CAMEO验证集的34.3%(31.5%)上,纯MSA(纯预训练)推理比预训练+MSA推理提供更好的TM-score。此外,纯MSA和纯预训练模式在不同的靶点上表现更好。如果我们总是对CAMEO数据集上表现最好的推理模式进行评分,整体的TM分数上升到85.0,预训练:MSA:预训练+MSA模式的贡献率总体上为25%:32%:43%。
创新点:
1.证明MSA表征比预训练模型信息量大,即使是最先进的生物预训练模型,如ESM-2,也不能轻易取代同源序列
2.证明基于预训练模型和基于MSA的模型都有独特的优势和劣势,构建了一个能够在多种模式下运行的架构,可以适应特定的环境(例如孤儿或快速进化的蛋白质的少MSA环境)。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢