
电子波函数计算是计算量子化学的一项基本任务。波函数参数的知识允许人们计算分子和材料的物理和化学性质。不幸的是,即使对于简单的分子,解析地计算波函数也是不可行的。
Hartree–Fock 方法或密度泛函理论 (DFT) 等经典量子化学方法允许计算波函数的近似值,但计算量非常大。降低计算复杂性的一种方法是使用能够以低得多的计算成本提供足够好的近似值的机器学习模型。
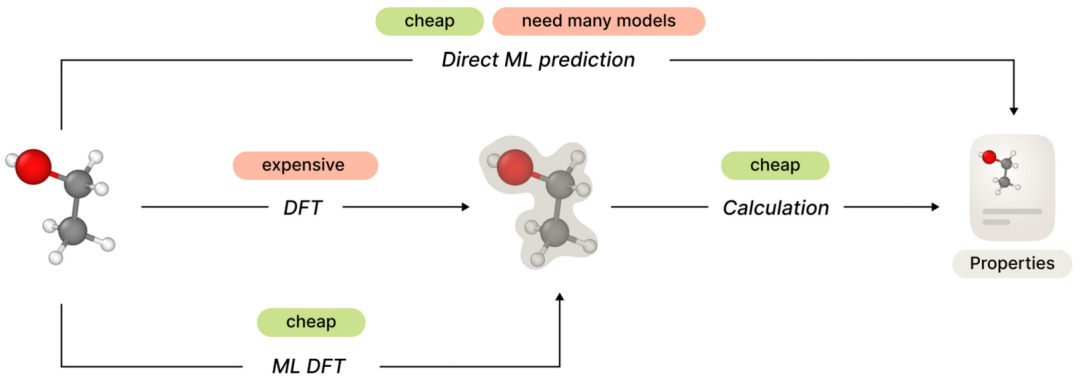
在最新的一项工作中,俄罗斯 AIRI 的研究人员介绍了一个新的精选类药物分子电子结构的大规模数据集,同时为多分子环境中分子特性的估计建立一个新的基准,并使用该基准评估各种方法。
研究表明,当从单分子设置切换到多分子设置时,最近开发的机器学习模型的准确性会显著下降。而且,这些模型缺乏对不同化学课程的概括。此外,这项工作提供的实验证据表明,更大的数据集会在量子化学领域产生更好的 ML 模型。
该研究以「nablaDFT: Large-Scale Conformational Energy and Hamiltonian Prediction benchmark and dataset」为题,于 2022 年 10 月 24 日发布在《Physical Chemistry Chemical Physics》。
论文链接:https://pubs.rsc.org/en/content/articlelanding/2022/CP/D2CP03966D
相关报道:https://phys.org/news/2022-11-world-largest-quantum-chemistry-dataset.html
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢