本文转自iTASSER公众号
蛋白质是一切生命系统的物质基础,它们密切参与着人体从触发免疫反应到大脑思考的每一个生理过程。蛋白质结构决定蛋白质功能,蛋白质只有正确折叠为特定的三维结构,才能发挥相应的生物学功能。我们想要从分子水平上了解蛋白质的作用机制,就需要精确测出蛋白质的3D结构。对于一个复杂蛋白质结构的生物学实验测定方法,往往需要耗费大量的时间和成本,甚至还不一定准确。历史上,动辄有科学家耗费几年、几十年时间才能得到一个清晰的蛋白质三维结构。
我们可以直接从氨基酸序列出发,利用计算机技术直接去预测蛋白质的三维结构吗?这一个问题也被称为蛋白质结构预测问题。蛋白质结构预测作为生物学里悬而未决的几大终极难题之一,近年来随着人工智能的发展,特别是深度学习方法的应用而曙光初现。2018年,谷歌 DeepMind 团队开发出的AlphaFold首次亮相便摘取CASP13的桂冠;在两年后的AlphaFold2版本更是在CASP14中,针对比赛中提供的大多数蛋白,预测准确性达到了可以与实验解析结构相媲美的精度。这一成就不仅获得了谷歌CEO皮猜、马斯克、李飞飞等大V纷纷点赞,就连马普所的演化生物研究所所长Andrei Lupas都直言“它会改变一切”。这一突破表明了人工智能对科学发现的影响,展示了人工智能在加速“解释和塑造世界基本法则”认识方面的巨大潜力。
在学术界欢迎鼓舞的同时,我们也需要清醒的认识到,和所有科技进步一样,AlphaFold2并不是十全十美的,它的表现并不是非常稳定。AlphaFold2在CASP14上只有2/3的蛋白预测做到实验精度,还有1/3并未做到,对于超级巨型的蛋白质复合体,对于蛋白质和DNA/RNA/小分子结合形成的复合物,预测能力和精度也还有待检验。2022年7月28日,DeepMind将AlphaFold蛋白质结构数据库从近100万个结构扩展到超过2亿个结构,几乎涵盖了所有基因组测序的生物体,彻底地改变了生物学研究范式。然而,在超过2亿个预测的蛋白质结构中,只有大约35%的结构具有高精度(按照AlphaFold2自己提供的模型精度评估方法估计)。
毫无疑问,AlphaFold2并不是一个完美的算法。虽然AlphaFold2本身提供了模型质量置信度指标,然而其模型可靠性除了生物学实验验证之外,目前尚且不能被外部解释或外部验证。学术界和工业届最终将产生许多改进版本,其中还有来自世界各地其他实验室开发算法与其竞争。就在刚刚结束的CASP15上,张阳实验室郑伟博士、山东大学杨建益开发的算法均超越了AlphaFold2的基准算法。这也意味着除了AlphaFold2本身自带的模型质量置信度度量之外,是否可能有一种第三方的模型质量评估方法能够从所有预测模型中选择出一个“最佳模型”?基于这样“最佳模型”,科学家们后续可以针对每个蛋白都设计出对应的分子进行调控(抑制或者激活),加速传统药物的研发过程的速度。有效的模型质量评估方法将会成为蛋白质预测结构(包括AlphaFold DB)实用性和推广性的关键;此外,模型质量评估方法对蛋白质结构精调和“最佳模型”识别等方面也具有重要意义。
浙江工业大学张贵军课题组于2022年12月2日在生物信息学领域期刊《Briefings in Bioinformatics》上在线发表了题为《Improved model quality assessment using sequence and structural information by enhanced deep neural networks》的研究论文(图1)。课题组基于之前发表的DeepUMQA算法(链接:Bioinformatics | 张贵军课题组:蛋白质结构模型质量评估方法DeepUMQA),进一步集成了蛋白质序列共进化信息特征、蛋白质家族结构特征,并设计了基于三角乘法更新规则和轴向注意机制改进深度网络的DeepUMQA2。CASP13和CASP14数据集测试结果表明DeepUMQA2性能分别比DeepUMQA提高了20.5%和20.4%("top 1 loss"指标);在三个月的CAMEO数据集(2022年3月11日至6月04日)上,DeepUMQA2大幅度优于DeepUMQA (‘AUC0,0.2‘指标),在CAMEO评测中连续三个月排名第一;在最近刚刚结束的CASP15中,DeepUMQA2的增强版本(DeepUMQA3)在复合物界面接触残基精度评估赛道中排名第一(链接:张贵军课题组在蛋白质结构预测大赛CASP15复合物界面接触残基精度评估赛道中斩获冠军)。尤为重要的是,“最佳模型”选择实验结果显示DeepUMQA2可以从AlphaFold2、RoseTTAFold、I-TASSER等蛋白质结构预测方法生成的众多模型模型中,识别出更好的“最佳模型”。该论文通讯作者为浙江工业大学信息工程学院张贵军教授,浙江工业大学信息工程学院刘俊博士生为第一作者。
图1. 《Briefings in Bioinformatics》发表论文
1. 研究方案
DeepUMQA2流程图如图2所示。对于要评估的结构模型,分别使用HHblits和HHsearch工具搜索多序列比对(multiple sequence alignment, MSA)和同源模板;然后提取MSA中的序列特征、模板中的结构特征和输入模型中的模型依赖特征,并将其连接成初始“对特征”;并将“对特征”通过基于三角乘法更新和轴向注意机制的神经网络进行迭代更新;最后通过两个独立的残差网络分别预测残基间距离偏差和阈值为15 Å的接触图,进一步用于计算模型每个残差的精度。
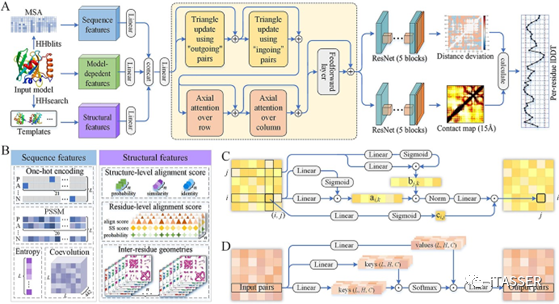
图2. DeepUMQA2概述。(A) DeepUMQA2的流程图;(B)从MSA中提取序列特征和从同源模板中提取结构特征;(C) ‘outgoing’三角更新规则;(D)轴向行注意力机制。
训练集和测试集构建:DeepUMQA2采用与DeepUMQA相同的训练集,包含7615个蛋白质,每个蛋白质包含大约150个模型。测试集为CASP13、CASP14和三个月CAMEO(2022年3月11日至6月04日)的数据集。CASP13数据集包含76个目标,每个目标约141个结构模型,共10739个模型;CASP14数据集包含70个目标,每个目标约有148个结构模型,共10738个模型;CAMEO数据集(2022年3月11日至6月04日)包含192个目标,每个目标约有10个结构模型,共有1882个模型。训练集中的蛋白质从Uniclust30 (Version 2018_08)中搜索MSA;从PDB100 (Version 03 March 2021)中搜索同源模板,并且删除2018年5月1日后发布的模板或序列恒等式大于30%的模板。对于CASP13和CASP14测试集中的蛋白质,MSA从Uniclust30 (Version 2018_08)中搜索;从PDB100中搜索同源模板,并分别删除2018年5月1日和2020年5月18日之后发布的模板。对于CAMEO测试集中的蛋白质,在UniRef30 (Version 2022_02)和BFD上搜索MSA,在PDB100上搜索同源模板。
2. 实验结果
蛋白质模型质量评估性能通过全局评价指标(Global QA)和局部评价指标(Local QA)两个方面评估。全局评估(Global QA)指标包括Pearson相关系数、'top 1 loss'、受试者工作特征(ROC)曲线面积AUC、假阳性率(FPR)阈值为0.2的修剪AUC0,0.2。Pearson相关系数显示了所有蛋白质模型的预测全局得分与实际全局得分之间的相关性,且相关系数越大,性能越好;'top 1 loss'是衡量根据评估分数挑选的最佳模型,与真实模型(由实验结构确定)的差异,差异越小 挑选最佳模型的能力越强。AUC和AUC0,0.2采用lDDT阈值0.6,用来区分好模型和坏模型(或残基)的能力。局部评价(Local QA)指标包括Pearson相关系数、平均逐残基的S分数 (ASE)、AUC和AUC0,0.2。ASE指标衡量逐残基平均S分数,评估性能随着ASE增加而增加;Pearson相关系数、AUC和AUC0,0.2与全局评价指标相似,区别在于它们是在局部残基上计算的。CAMEO竞赛根据局部AUC0,0.2对模型质量评估方法进行默认排名。
(1) CASP13和CASP14数据集测试性能
在CASP13和CASP14数据集上测试DeepUMQA2的性能,并与目前最先进的方法进行比较,包括ProQ2,ProQ3,ProQ3D,ProQ3D-lDDT,ProQ4,VoroMQA, 3DCNN,ModFOLD7,ModFOLD8,GraphQA,QDeep,Ornate,QMEANDisCo ,DeepAccNet和DeepAccNet-MSA。所有比较方法的数据均来自CASP官方网站(https://predictioncenter.org/download_area/)的存档数据。DeepUMQA2和比较方法在CASP13和CASP14数据集上的性能分别如表1和表2所示。
在全局评估基准方面,无论是CASP13还是CASP14数据集上,DeepUMQA2在所有指标上都优于所有同类方法。DeepUMQA2在CASP13和CASP14数据集上的全局Pearson相关系数分别为0.919和0.899,比次优方法ProQ3D-lDDT和DeepAccNet-MSA分别高8.1和4.9%。这一结果表明DeepUMQA2能够准确地捕捉模型的全局精度。DeepUMQA2在CASP13和CASP14数据集上的“top 1 loss”分别为0.049和0.035,优于所有同类对比方法。DeepUMQA2的“top 1 loss”比DeepAccNet-MSA的“top 1 loss”低10.3%,在CASP14中达到了最低的“top 1 loss”。这一结果表明,该方法能够更好地从大量的预测模型中识别出最佳模型。为了分析DeepUMQA2和对比方法区分好和坏模型的能力,基于所有模型的预测全局精度与实际全局精度进行了ROC分析。图3A和图3B分别显示了不同方法在CASP13和CASP14数据集上的ROC曲线。DeepUMQA2在CASP13和CASP14数据集上的AUC分别为0.982和0.961,显著优于对比方法。DeepUMQA2的AUC0,0.2优于所有在CASP13和CASP14数据集上的对比方法。
在局部评价基准方面,DeepUMQA2在CASP13和CASP14数据集上仍然是所有方法中表现最好的。DeepUMQA2在CASP13和CASP14数据集上的局部Pearson相关系数分别为0.868和0.822,比次优方法ProQ3D-lDDT和DeepAccNet-MSA分别高11.0和7.5%左右。DeepUMQA2在CASP13和CASP14数据集上的ASE分别为0.915和0.910,显著优于所有对比方法。结果表明,该方法预测的残基精度总体上更精确。基于所有模型残基的预测和实际精度的ROC曲线如图3D和3E所示,在CASP13和CASP14数据集上,DeepUMQA2在所有方法中AUC、AUC0,0.2最高。这一结果表明DeepUMQA2预测的局部精度分数可以区分结构模型中的准确残差和不准确残差。为了进一步分析DeepUMQA2和对比方法的综合性能,分别使用所有全局评价指标和所有局部评价指标的z-score得分之和对这些方法进行了排名。图4给出了所有方法在全局和局部精度估计方面的综合表现。可以发现,在CASP13和CASP14数据集上,DeepUMQA2的全局和局部综合性能是所有对比方法中最好的。
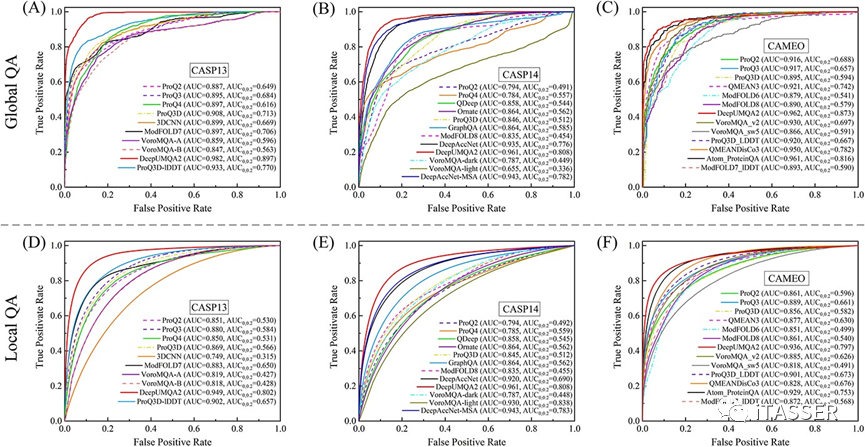
图3. (A) CASP13、(B) CASP14和(C) CAMEO数据集上全局评价指标(Global QA);(D) CASP13、(E) CASP14和(F) CAMEO数据集上局部评价指标(Local QA)。其中lDDT截断值为0.6,在Global QA中用来区分高质量模型和低质量模型,在Local QA中用来区分高质量残基和低质量残基。
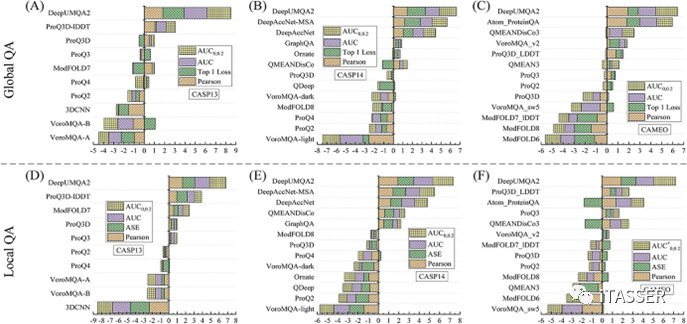
图4. 全局和局部准确性评估方法的排名(Global QA和Local QA)。(A-C)分别根据CASP13、CASP14和CAMEO数据集上所有Global QA指标的z-score进行排名。(D-F)分别根据CASP13、CASP14和CAMEO数据集上所有Local QA指标的z-score进行排名。
(2) CAMEO上的测试性能
为了进一步验证DeepUMQA2的性能,DeepUMQA2连续三个月(2022年3月11日至6月04日)参加了CAMEO-QE (https://www.cameo3d.org/quality-estimation/3-months/quality/all/)的盲测,在所有参与的模型质量评估(MQA)方法中排名第一。在三个月的CAMEO数据集上,论文详细分析了DeepUMQA2和其它参赛服务器方法的性能,其中所有模型评价数据均来自CAMEO官网。对比方法包括Atom_ProteinQA,QMEAN3,QMEANDisCo3,ProQ2,ProQ3,ProQ3D,ProQ3D_LDDT,ModFOLD6,ModFOLD7_LDDT,ModFOLD8,VoroMQA_v2,VoroQA_sw5。
所有方法在全局和局部指标上的对比性能如表3所示。在全局评价指标方面,DeepUMQA2的Pearson相关系数为0.899,优于其他方法;DeepUMQA2的“top 1 loss”为0.017,仅次于Atom_ProteinQA;DeepUMQA2的AUC和AUC0,0.2最高,DeepUMQA2的AUC0,0.2指标显著高于其他方法,比次优方法Atom_ProteinQA高约7.0%。在局部评估指标方面,DeepUMQA2在所有性能指标上优于所有其他方法。DeepUMQA2的Pearson相关系数和ASE分别为0.870和0.910,比排名第二的Atom_ProteinQA (Perason: 0.829)和ModFOLD7_lDDT (ASE: 0.858)分别高4.9和6.1%左右。值得注意的是,DeepUMQA2的局部AUC0,0.2为0.797,显著高于其它方法,比次优方法Atom_ProteinQA高出10.8%左右。CAMEO官方用局部AUC0,0.2对MQA方法进行了排名。分别使用所有全局评价指标(图4C)和局部评价指标(图4F)的Z-score对所有方法进行排名,可以发现DeepUMQA2在Global QA和Local QA中综合表现最好。
(3)组件实验
组件实验结果表明,序列特征、结构特征和基于注意力机制的网络架构对DeepUMQA2而言都是性能影响因素。
(4)案例分析
图5给出了对CASP14目标T1034的定量分析,该目标包含145个预测模型,真实的全局lDDT分布在[0.582,0.752]。图5B显示了这145个模型上真实的全局lDDT和DeepUMQA2预测的全局lDDT的比较,结果显示它们相关性很高,Pearson相关系数为0.917,R2为0.841,根据预测的全局lDDT选择的“最佳模型”为真实的最佳模型。图5C绘制了全局和局部ROC曲线,全局AUC和AUC0,0.2均为0.993,局部AUC和AUC0,0.2分别为0.947和0.756,ASE为0.945。图5D-5F显示了三种不同精度模型的结构,其对应的预测和真实的lDDT,以及每个残基的预测和真实的lDDT得分。可以发现,DeepUMQA2预测的每个残基lDDT能够捕捉残基精度变化趋势,且易于区分残基的高、低精度区域,为模型的进一步细化提供有利信息。
图5. DeepUMQA2对CASP14目标 T1034质量评估性能的定性分析
(5) “最佳模型”识别实验
DeepUMQA2可以用于选择主流蛋白质结构预测方法的“最佳模型”。论文从CASP14官网收集了AlphaFold2预测的66个目标和I-TASSER预测的70个目标的前5个模型。此外,他们还从CAMEO官网收集了RoseTTAFold在2022年3月11日至6月04日三个月内预测的182个目标的前5个模型。分别对这三种方法预测的结构模型进行评估,并从中选择评估得分最高的模型与这些方法给出“最佳模型”进行比较。表5给出了与AlphaFold2、I-TASSER和RoseTTAFold的比较结果。
在AlphaFold2的66个目标上,DeepUMQA2选择的“最佳模型”TM-score平均值为0.866,高于AlphaFold2的“最佳模型”TM-score平均值0.859。DeepUMQA2在68个目标中选择了26个“最佳模型”,AlphaFold2选择了21个“最佳模型”。DeepUMQA2选择的“最佳模型”在31个目标上比AlphaFold2的选择的“最佳模型”更准确,AlphaFold2的“最佳模型”在16个目标上更准确。对于I-TASSER的70个目标,DeepUMQA2选择的“最佳模型”TM-score平均值为0.672,比I-TASSER的“最佳模型”的TM-score平均值(0.656)提高了2.44%。DeepUMQA2在70个目标上挑选出43个“最佳模型”,比I-TASSER的“最佳模型”(31个)高出12个。DeepUMQA2选择的“最佳模型”在28个目标上比I-TASSER的“最佳模型”更准确,I-TASSER的“最佳模型”在7个目标上更准确。在RoseTTAFold的182个目标中,DeepUMQA2选择的“最佳模型”的TM-score平均值为0.837,高于RoseTTAFold “最佳模型”的TM-score平均值0.835。DeepUMQA2在182个目标中选择了69个“最佳模型”,RoseTTAFold选择了50个“最佳模型”。DeepUMQA2选择的“最佳模型”在68个目标上比RoseTTAFold的“最佳模型”更准确,RoseTTAFold的“最佳模型”在31个目标上更准确。可以发现,DeepUMQA2可以从AlphaFold2, I-TASSER或RoseTTAFold预测的模型中选择出更为准确的“最佳模型”。
3. 结论
单链蛋白质结构的预测已取得重大突破,但是复合物结构的预测仍有很大提升空间。如何准确评价复合物结构模型质量,尤其是蛋白质相互作用界面接触残基的质量,有望能够为提高复合物结构的预测精度提供一种重要的手段。
该研究得到了科技部2030-“新一代人工智能”重大项目(2021ZD0150100)、国家自然科学基金项目(62173304)、浙江省自然科学基金重点项目(LZ20F030002)的资助。
附
DeepUMQA2论文链接:https://doi.org/10.1093/bib/bbac507
DeepUMQA2服务器:http://zhanglab-bioinf.com/DeepUMQA2/
内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢