
分享嘉宾|张清鹏 香港城市大学 副教授
编辑整理|熊丹妮 恒瑞医药出品社区|DataFun
AI 在医药领域、药物发现等方面的应用是一个相当热门的研究领域。本次会议将介绍的研究和这些高度相关,不过涉及分子结构、蛋白质结构的内容会比较少,更多关注如何结合临床进行应用。今天主要将介绍如何结合网络科学和机器学习(GCN算法)对多种抗癌药物的联用进行预测。
本团队做了很多网络科学相关的工作,在健康领域主要做的是临床数据相关以及传染病的研究,以及生物网络的研究。今天将给大家介绍一篇团队去年发表的第一篇生物网络相关研究,本项工作的合作者是博士生羊剑楠,徐仲之,香港中文大学医学院 William Wu 教授,以及华中科技大学附属同济医院的褚倩教授。
本文主要内容:AI 药物发现与再利用背景介绍,GraphSynergy 模型介绍,和后续工作。
01
AI 药物发现与再利用背景介绍
首先简单介绍一下 AI 药物发现与再利用的背景。
1. AI 药物发现与再利用

左侧图中可以看到,第一个完全由 AI 研发的药物在今年年初进入了临床试验阶段,月初该公司(Insilico Medicine)的第二个 AI 药物(新冠相关)也进入了临床试验。Insilico 总部在香港科技园(主要团队成员一个是中国人,一个是俄裔美国人),Insilico 的很多工作都非常领先,是该领域的独角兽,有足够的创新能力。
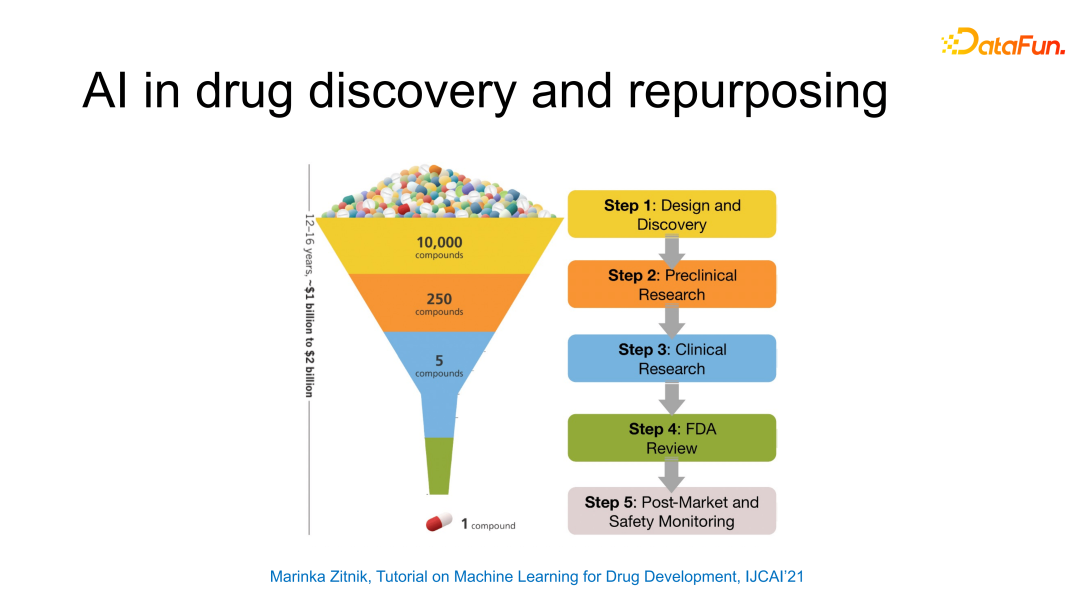
这张图片主要揭示了研发一个药物的难度及成本,在每一个药物研发都要花费 10-20 亿美元的情况下,如果在其中的任何一部分能得到一定的加速/辅助,都会是一个非常有影响力的研究成果。
2. 面临挑战
在这个研究领域,本团队所关注的不是如何发现新的分子/蛋白质结构来作为新的药物,而是“老药新用”(drug repurposing),以及现有药物如何进行联用来得到更好的效果。

抗癌药物的联用大家可能有一定的了解,“是药三分毒”,癌症药物尤其突出,很多癌症病人最终可能是因为癌症药物的强烈副作用去世的。另一方面,癌症药物的组合往往能带来更好的抑制肿瘤的效果,所以研究者们想知道当 2 个或 者 3 个药物结合在一起联用时,是否能得到“1+1>2”的效果。现有研究中有从机器学习角度的,也有网络科学角度的,这类研究中第一个存在的问题是它们大多数都是黑匣(black box)模型,解释性不够强;第二个问题是已有模型更多关注化合物的分子结构,如折叠方式、角度等。
所以之前研究的假设一般是:两种药物有相似的化学结构,它们可能就会有相似的治疗效果,以此为依据再来预测它们联用对于某些癌细胞的抑制效果。这些研究存在的一个比较大的问题是,忽略了人体蛋白质之间非常丰富的生物交互关系,如药物和癌细胞都会影响到一部分的蛋白质,这些蛋白质在人体的蛋白质交互网络中的结构关系是怎样的,这类问题研究得不够深入。
3. 理论基础

另一方面,在网络科学领域也有一些相关进展。网络科学的研究范式很多是统计物理,故都是以一些显性公式来尝试描述药物和疾病之间的关系。一般来说并不一定是最优的结果,但是结果的解释性很强。本研究的基础是美国东北大学 Barabasi 博士团队发表的 2 篇文章,在文章中集中研究了药物和癌细胞所影响的蛋白质的结构关系:人体内的蛋白质交互网络可以作为人体机能的骨骼式架构。虽然不同的病人有自己独特的临床表征,但是所有人种的蛋白质交互关系大致是相似的。

如上图中所展示的,粉红色是癌细胞所影响的人体蛋白质,黄色和绿色分别代表了两种抗癌药物的靶点蛋白质,文章中用了非常显性可解释的方式来提出方法,文章的假设是:如果抗癌药物的靶点正好是癌细胞所影响到的蛋白质(颜色有重合),那么该抗癌药物可能会有比较好的治疗效果。对于多种药物联用的情况,文章的假设是:如果两个药物有互补效应,即都可以覆盖到被癌细胞影响的蛋白质,且覆盖的蛋白质不一样,这就是最优的情况。如果两种药物(绿色和黄色)作用的蛋白质重合,这样虽然是有效的但是存在过度暴露(over exposure)的情况。即使用了两种药物,但它们作用的蛋白质是同样的,要承受两种药物所带来的毒性,故属于不是非常好的药物联用方式。
基于这些假设,文章提出了一些方法来定量的研究这些子图的关系。如计算距离时假设它们服从一个分布(如正态分布),来计算红圈和绿圈之间的距离,节点之间随机选择的,通过概率的方式来计算它们是否足够接近。文章也提出了一些新的方法,如 separation score。距离的计算方法可以有很多,大家也可以提出自己的计算方法,来衡量红圈和绿圈的距离,如重合node的数量,红圈任意 node 到绿圈任意 node 的最短路径,不同的 node-pair 可以算平均/最短/最长。本质上来说这两篇文章所发表的方法是非常显性的计算方式,非常显性的表示网络邻近性(Network Proximity),其实就是通过网络的拓扑结构来衡量子图之间的关联,在此基础上来预测药物联用效果。

这类方法的思路非常直接并且具有很好的可解释性,但是它也存在一些问题。
首先,它基本关注拓扑结构,但实际上网络中的结构相当丰富,如果仅用拓扑距离会缺失一定的精度,尤其是不同蛋白质间存在着非常复杂丰富的交互关系,如果只考虑最短路径是无法覆盖这些交互特征的。
其次,现有研究中的网络科学方法认为这些不同蛋白质是同质的,但实际上不同蛋白质对于抑制肿瘤发展的贡献是不一样的。
第三个问题是这种显性的公式来计算药物可能的机制是比较死板的,无法进行下一步优化,很可能不是最优的结果。
02
GraphSynergy 模型介绍
基于前面提到的一系列问题,本研究提出了 GraphSynergy 方法。
1. 模型框架

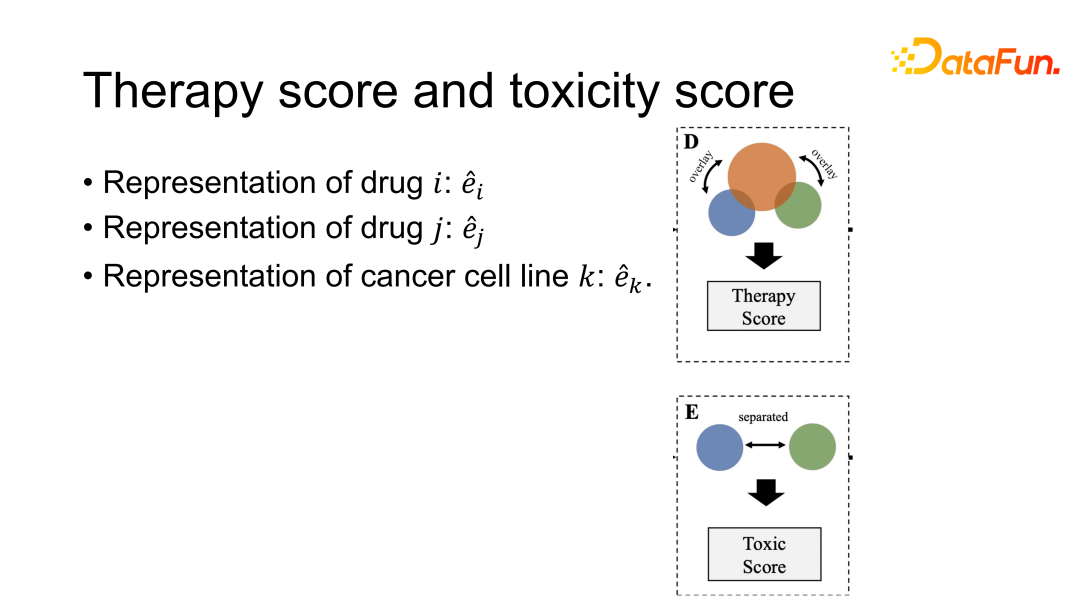
GraphSynergy 方法是在癌细胞中,预测不同抗癌药物的作用机制,以及对人体蛋白质交互网络的影响。GraphSynergy 使用了网络科学的两个假设:一个是通过 Therapy Score 来计算癌症药物治疗癌症的效果,以及联用的效果;另一个 Toxicity Score,用于评价两种药物联用的毒性效应,主要是计算两种药物 overlap 的程度,如果 overlap 比较高,则认为两种药物的过度暴露风险就会较高,带来的毒性也会比较多。最终在 Therapy Score 和 Toxicity Score 之间找到一个平衡。
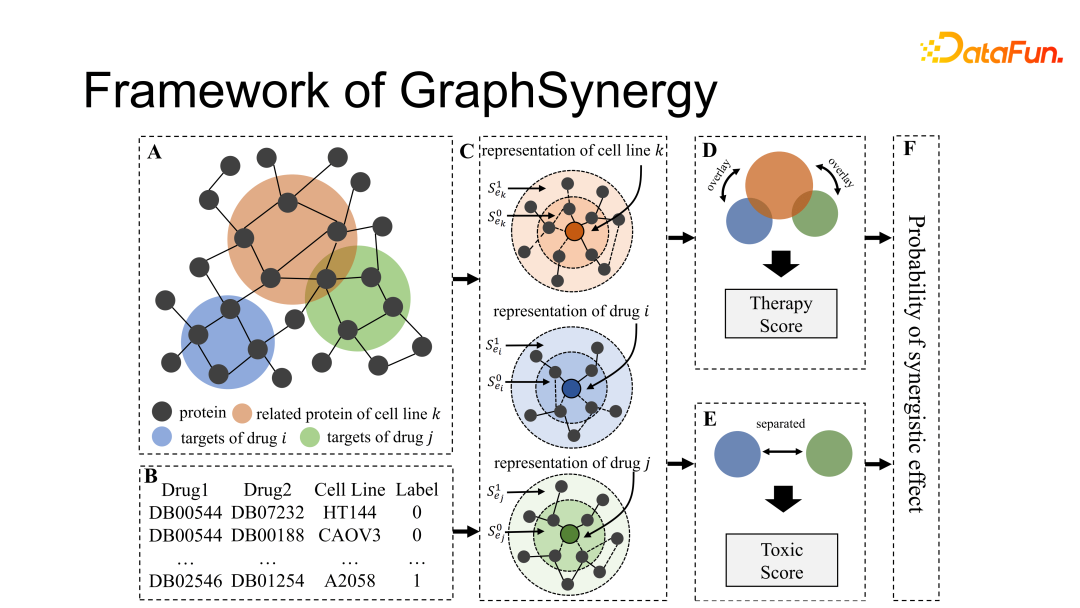
上图展示了 GraphSynergy 的主要框架。
图 A 中黑色的点,即不同的 node 代表了人体中不同的蛋白质,这些蛋白质间的交互关系就是图中的边(edges),红色、蓝色、绿色分别代表了癌症所影响的蛋白质,药物 i 和药物 j 的蛋白质靶点。
图 B 展示了存储在数据库中进行训练和验证的数据:三元组(药物1,药物2,所对应的癌细胞系编号)及 label(是否在数据库被记录为有积极的治疗效果)。模型主要强调的是节点的局部特征(local properties),即它和周围蛋白质间的交互关系。
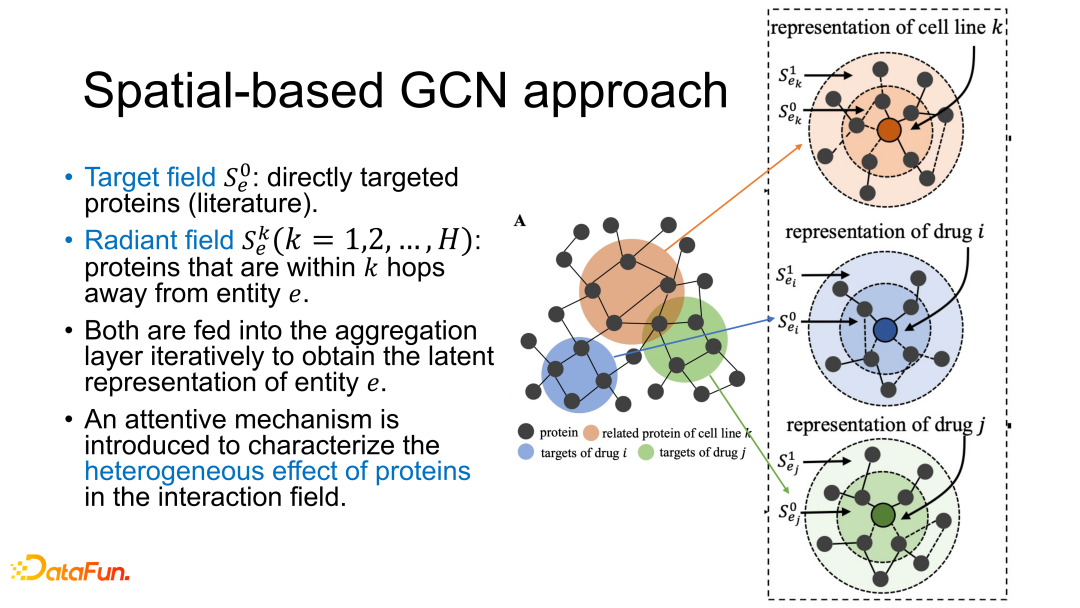
C 图中多加的红点代表癌细胞,与红点相连的内圈节点代表了癌细胞直接影响的蛋白质,这个称为 target field(直接被影响的蛋白质,现有研究主要研究这部分蛋白质间的关系),外圈节点是 target field 的延伸,称为 radiant field,是直接相连的蛋白质节点的 n 跳邻居节点(n 跳需要通过实验计算确定)。
最终通过上面的表示方式来计算得出 Therapy Score 和 Toxic Score,并将两个得分进行整合,最后计算这两种药物针对这种癌症联用效果的概率。
2. 模型原理解析

具体来讲,GraphSynergy 主要涉及到了邻域聚合(Neighborhood aggregation)方法。首先使用基于空间的 GCN 模型对所有节点进行初始 embedding,然后对给定的实体(药物/细胞系)及其交互域(直接相关的蛋白质)中的每个蛋白质都给一个贡献权重(contribution weight)(其中g是向量内积),通过 attention 机制来学习权重,最后每个蛋白质的权重经过归一化处理后,会对当前层再进行加和计算。
接下来在聚合层会把蛋白质的影响聚合到一起,在每一层都会进行聚合处理,分别计算每层内的蛋白质贡献,再把不同蛋白质的贡献聚合到一起,这样就有 n 个 embedding 向量来代表 n 层。其中 标记为的是目标域, 是直接相关目标域的层(目标域向外延伸了一层),这样向后延伸直到 H 层,最后将 H 层向量直接拼接到一起(w,b 为模型参数)。

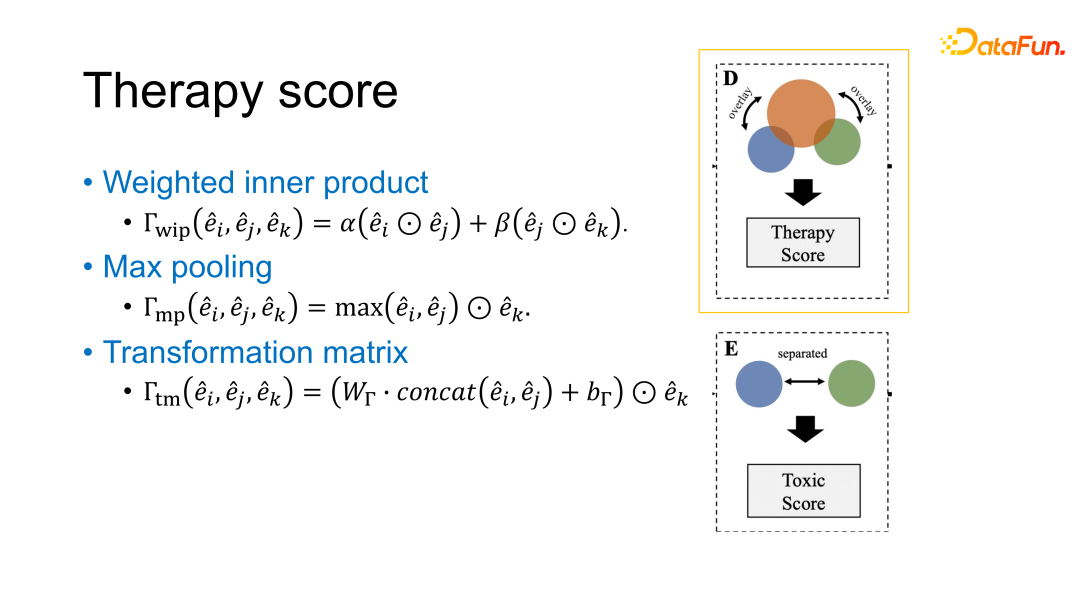
在计算治疗得分时,尝试了以下三种方法:
-
加权内积:分别计算药物 i、j 与癌细胞系 k 的相似度,最后取一个加权和/均值;
-
最大池化:将两种药物的 embedding 做一个最大池化,然后再计算它们和癌细胞系之间的关系;
-
转移矩阵:将两种抗癌药物的 embedding 拼接到一起,然后再计算它们与癌细胞系 embedding 之间的关系。
毒性得分的计算相对简单,假设是两种药物越相似,它们过度暴露的问题会越重,带来的额外毒性会更强,所以直接计算两种药物的相似度,以它来指代这两种药物联用的毒性得分。

最后计算治疗得分和毒性得分的差值后再过一个 sigmoid 激活函数,得出这两种药物联用对于特定癌细胞是否有效的预测(loss 函数使用的是二元交叉熵)。
3. 数据集

用于实验的数据集有以下三个:
-
PPI 网络(protein-protein interaction network):15970 个不同蛋白质间的217160 种交互关系。
-
药物蛋白质靶点关系(drug-protein associations):4428 种药物和 2256 种人体蛋白质间的 15051 种关系。
-
癌细胞系和蛋白质间关系(cancer cell line-protein associations):PPI 网络中 18022 种基因所编码的蛋白质,1035 种癌细胞系,以及 749551 种关联关系。
用于训练和预测的数据集整合了 DrugCombDB 和 Oncology-Screen 两个数据集。其中 DrugCombDB 中药物有 764 种,细胞系有 76 种;Oncology-Screen 中药物有 21 种,细胞系有 29 种。但是数据库中的三元组合关系会有很多种,如 DrugCombDB 中正向关系有 31623 种,负向关系有 37813 种。
4. 模型基线

进行模型验证时参考了以上 7 种基线模型:
-
Network Proximity(NP):纯网络科学机制,计算网络中的 z-score 和 separation score,然后通过公式计算不同子图之间的网络临近,从而计算药物联用的效果;
-
GraRep:基于矩阵分解的方式;
-
DeepWalk 和 Node2Vec:图计算中经典的边预测和知识表示方式;
-
DeepSynergy:基于传统 DNN 框架;
-
GCN和KGNN:基于空间的 GCN 和基于 GNN 的预测模型 KGNN。
上表总结了 GraphSynergy 的预测效果:GraphSynergy 在两个数据集上的效果都优于之前的基线模型。NP 模型的效果比较差且和其他模型效果间的差距也很大,其他模型间的差异不是特别大,但 GraphSynergy 的效果有显著提高。
5. 模型变种
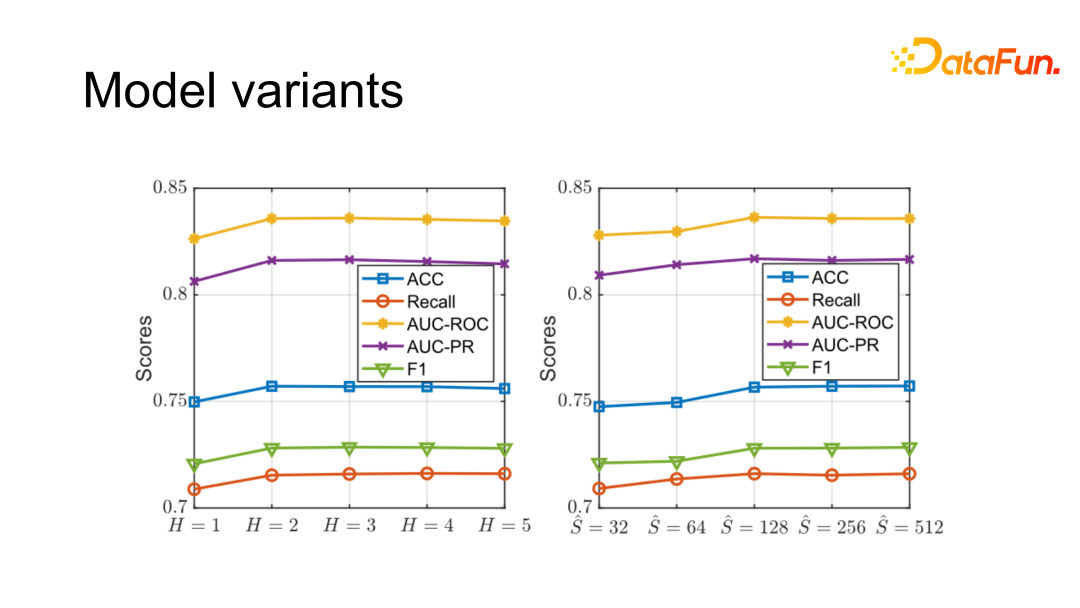
对于不同的模型变种也进行了分析,如目标域的辐射范围(辐射域层数)是可以变化的,如果辐射太少模型考虑到的局部特征就会变少,辐射太多会使信息太综合且增大了计算量。如上图所示,辐射到 2 层就能得到比较好的结果,再往后提升效果就没有很明显计算量也会增大,故选择 H=2 的模型结果。
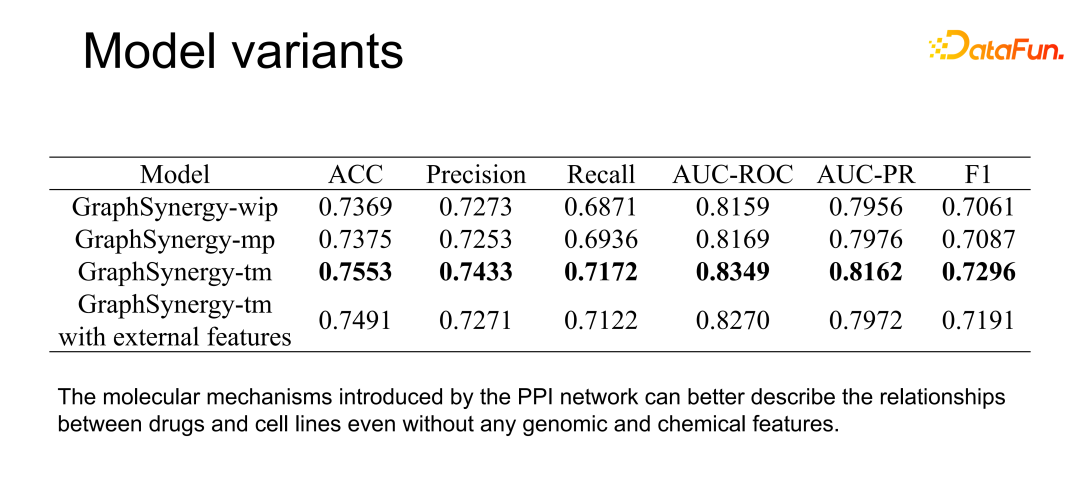
前文提到,在得到 embedding 后需要进行整合,对于不同的整合方式也进行了探究。上表可以看出,直接将两种药物的 embedding 整合到一起,再计算和癌细胞系 embedding 间的关系,这种方式的效果相对来说最好。业内也有一些基于基因组和化学结构的算法来计算药物联用效果,故尝试了在模型中添加基因组和化学结构特征,但实验结果发现模型效果并未得到提升。说明模型仅通过蛋白质交互网络的信息已经可以达到非常好的预测效果了。
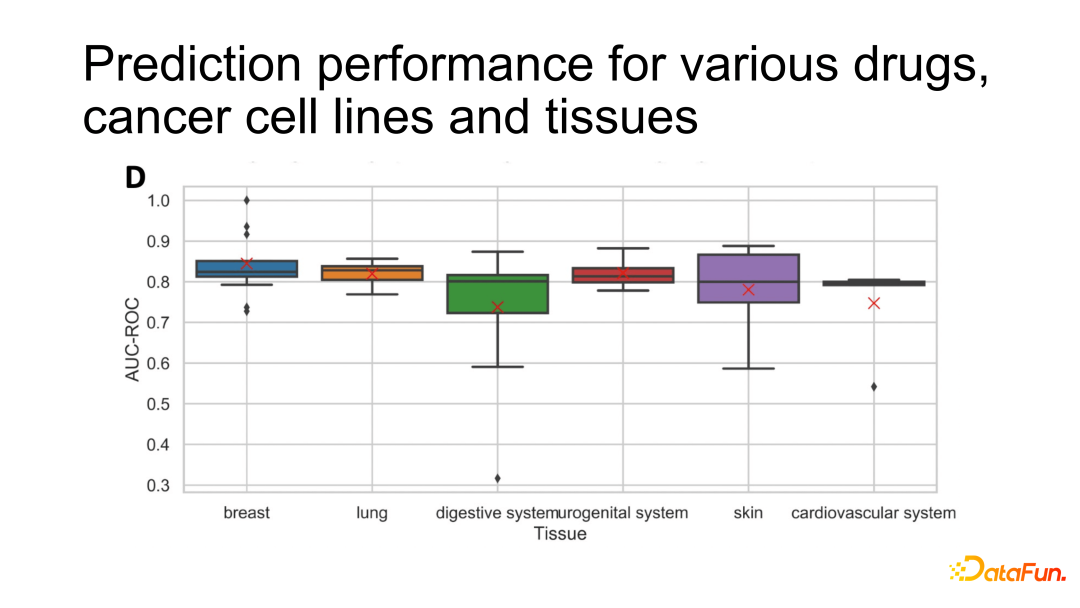
通过回溯分析,对模型针对不同癌细胞系的预测效果进行了探究。总体来说 AUC 都比较高,模型对于乳腺癌、肺癌、泌尿系统癌症的预测效果是最好的,并且方差也比较小;对于皮肤癌、消化道系统癌症的预测效果虽然不错,但是方差比较大;对于心血管癌症来说,可能由于数据量比较小,分布不是非常显著。
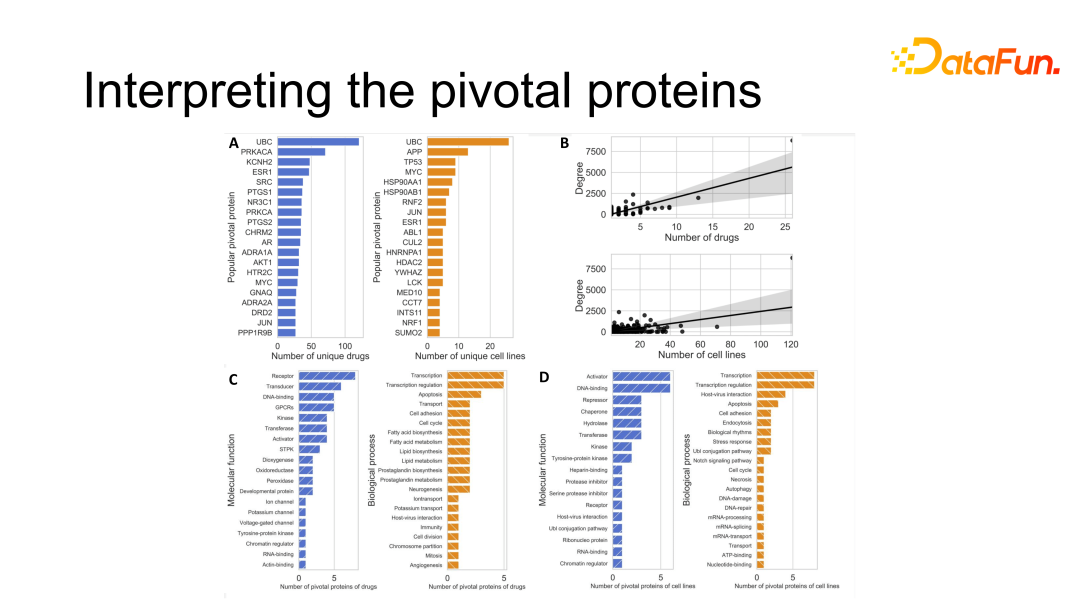
如上图所示,基于模型中的 attention 机制,我们计算了比较重要的蛋白质,即两种药物联用治疗一种癌细胞的机制中比较重要的人体蛋白质,对这些蛋白质进行了分析。对与治疗效果相关度比较高的蛋白质进行了排序,然后对比了这些蛋白质分子功能和生物功能的关系。
总结如下:

从分子功能上来看,与药物相关的重要蛋白质在人体中主要承担了受体(receptor)和转导体(transducer)的角色,与癌细胞相关的蛋白质则主要作为激活物(activator)和 DNA 结合物(DNA-binding)。简单来讲,激活物和 DNA 结合物是控制癌细胞生长的关键蛋白质,而药物则是通过受体和转导体来施加效果以抑制癌细胞生长。
在生物过程中,与药物和癌细胞相关性比较高的蛋白质都是和转录以及转录调控过程相关的蛋白质。
6. NSCLC 案例介绍
为了增加可解释性,下面将通过一些例子来看看我们所得出的结果和之前网络科学所得到的结果间的异同。
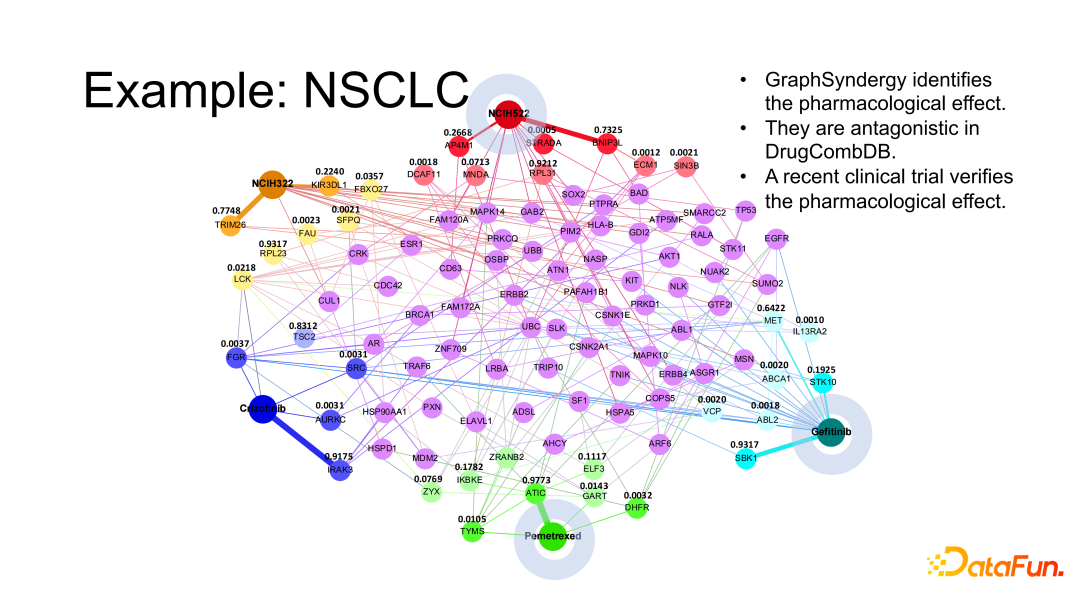
如上图所示,我们找出了 3 种药物(Crizotinib、Pemetrexed、Gefitinib)以及 2 种肺癌的癌细胞种类(NCIH322、NCIH522),其中 Pemetrexed 是常见的化疗药物,Crizotinib 和 Gefitinib 是两种靶向药物。图中可以看出,Pemetrexed 和 Crizotinib 联用对 NCIH322 的治疗效果比较好,而 Pemetrexed 和 Gefitinib 联用对 NCIH522 的治疗效果比较好。实际在数据库中并未有 Pemetrexed 和 Gefitinib 联用治疗 NCIH522 的记录,但是算法计算出来这种联用方式是有效的。通过查阅发现最近的一项临床实验验证了 Pemetrexed 和 Gefitinib 的联用机制,可能不久之后它们也会被 FDA 批准作为治疗 NCIH522 的一种方案。这体现了算法发现新的药物使用方式的潜力。

前文中提到的网络科学方法比较强调有蛋白质间的重叠,但 GraphSynergy 网络中的药物靶点和癌细胞直接相关的蛋白质间并没有直接的重叠。GraphSynergy 是通过非常丰富的局部链接最终对癌细胞起到抑制作用,也说明我们需要考虑这些靶点之间的局部子图关系,以及局部目标蛋白质以什么方式影响癌细胞所影响的蛋白质。通过 GraphSynergy 发现的一些比较重要的蛋白质与最新生物医学文献证据的吻合度也很高。
7. 结论
最后总结如下:

以网络科学的角度来探索药物联用问题,将网络科学和 GCN 框架结合到一起得到更优的抗癌药物联用机制算法,通过对治疗效果和毒性效果的考量,最终得到的算法相较于以前的基线预测效果能有显著的提升。
03
后续工作
1. 多层生物网络
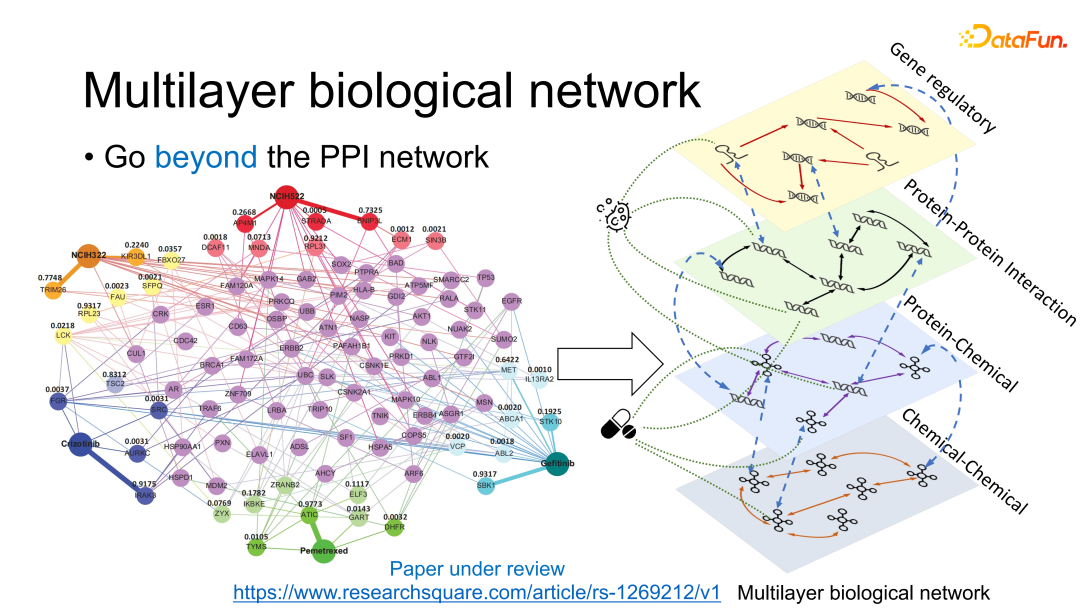
在以上工作的基础上,我们认为如果只看蛋白质交互关系可能过于简单,所以将单层的蛋白质交互网络进一步扩展为多层的人体生物网络,继而开发了一个新的解释性更强的算法。
2. 可解释性
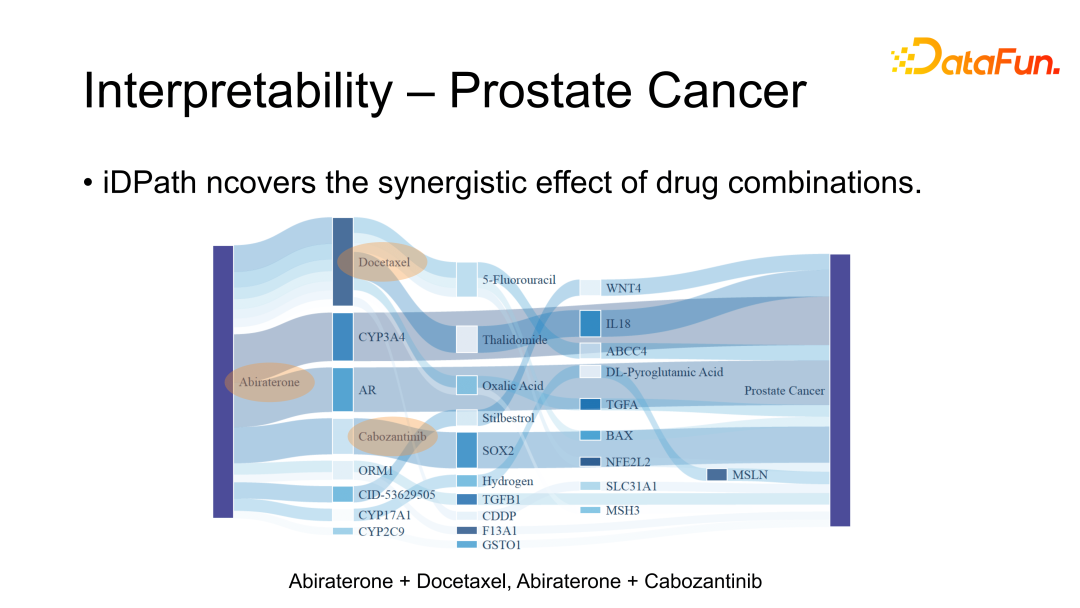
这个算法能够给出一个显性通路来指出不同药物对不同癌细胞的作用方式,即可以将药物作用机制(mechanism of drug action)显性的学习出来。通过药物反应机理的通路可以发现药物联用的可能性,以及不同药物的再利用方式。
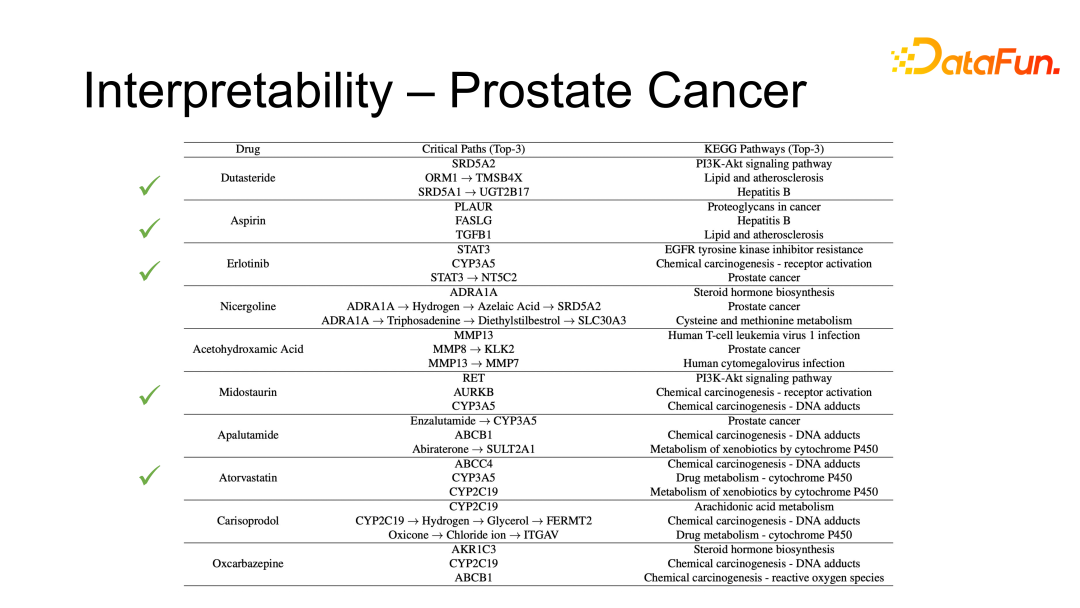
以前列腺癌为例,以上表格展示了新模型所发现的 10 种可以治疗前列腺癌的药物,其中有 5 种已经被 FDA 批准使用了。
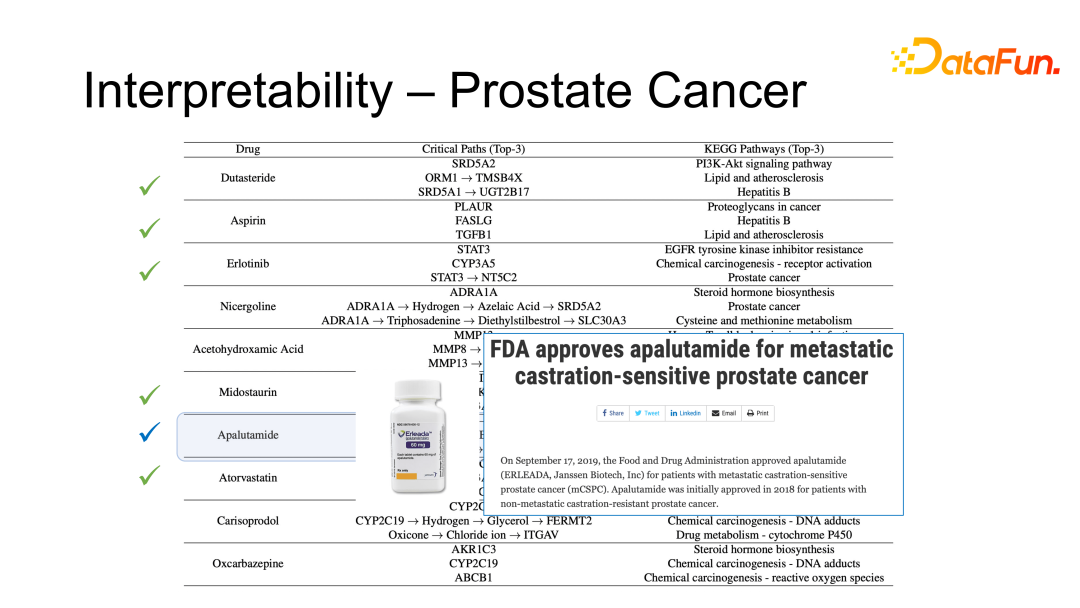
有 1 种在数据库中尚未收录,是 FDA 2019 年刚刚批准的,即算法发现了一些刚刚被批准但尚未记录在案的药物,侧面反应了算法的合理性。
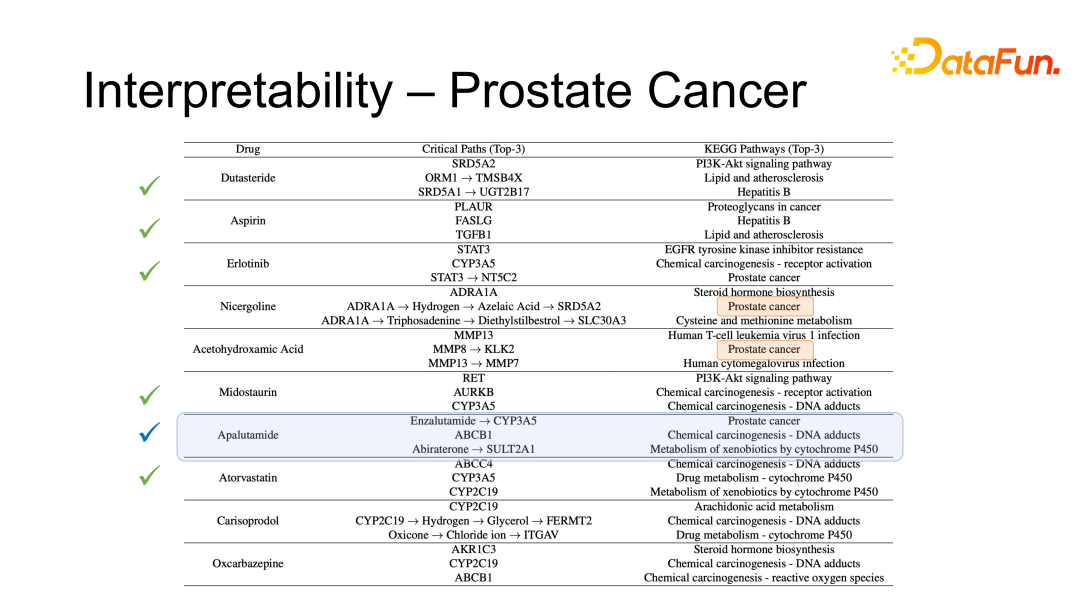
剩下的 4 种药物,有两种药物在 KEGG 生物通路中与前列腺癌的相关性也很高。在香港大学医学院汇报时,药学系同学反馈他们也在研究这两种药物是否可以治疗前列腺癌,故这两种药物也有可能被用于治疗前列腺癌。
通过这样的验证,提示我们算法是可以发现一些药物的新用法以及药物联用方式。
参考文献:
[1] Yang, J.†. , Xu, Z.†. , Wu, W.K.K. , Chu, Q. & Zhang, Q.*. (2021). GraphSynergy: Network Inspired Deep Learning Model for Anti–Cancer Drug Combination Prediction. Journal of the American Medical Informatics Association. 28/11. 2336 - 2345. doi:10.1093/jamia/ocab162
[2] Yang, J.†. , Li, Z. , Wu, W.K.K. , Yu, S. , Xu, Z.†. , Chu, Q.*. & Zhang, Q.*. (Nov 2022). Deep Learning Identifies Explainable Reasoning Paths of Mechanism of Action for Drug Repurposing from Multilayer Biological Network. Briefings in Bioinformatics. 23/6. bbac469 doi:10.1093/bib/bbac469
04
问答环节
Q1:针对多层生物网络,之前是在一张图上考虑所有的交互关系,后面有做不同的分层,层与层之间以及层内的关系是如何进行建模的?如何对这些数据库进行对齐?
A1:我们将多个数据库进行了整合,主要利用了层与层之间的关系,层之间的关系,如基因调控到蛋白质,蛋白质与不同的化合物之间等是通过数据库将它们链接到一起,这其中整合了相当大的一些数据库。以蛋白质和化合物之间的关系为例,是有数据库专门描述蛋白质与化合物之间的关系的;以基因调控与蛋白质交互之间为例,基因与蛋白质之间的转录关系也是直接有数据库的,故是通过公开的数据集对它们进行整合的。当然其中也是存在遗漏的部分,生物医学本身数据就做不到很全面,我们只能尽力去将每条边从数据库中进行标记。
Q2:未对齐的数据对模型效果的有影响吗?或者在什么比例下会对模型效果有比较大的影响?
A2:我们的研究中尚未考虑这个问题,我们的研究假设是人体蛋白质网络就是目前的结构。
Q3:异构图是如何进行计算的?
A3:模型中有不同的层,我们在计算时也考虑了不同的机制(详情可以查看文章的预发表链接)。我们直接将其转成异构网络,再做 embedding;也可以分层处理再整合到一起。在我们的研究中,想强调模型的可解释性,故先从网络拓扑分析着手寻找可能的生物通路,在多层网络上做 embedding 之后再进行计算预测。算法是先发现通路,通路上有不同层的节点,再将不同层的节点整合到一起,最终生成结果。
Q4:在图比较复杂时,如何抽取某个蛋白质的子图进行分析?
A4:我们尚未对这方面的子图分析进行研究。我们的训练目的和数据都是基于某种药物是否能对某种癌细胞进行治疗,并且是以通路的形式进行预测,包含了更丰富的信息。研究的主要目的是寻找药物和疾病之间的通路,并未对子图进行深入研究。
Q5:网络的每层特征都是不同的,如何对这些特征进行整合?
A5:针对这个问题,我们做了一些研究假设,如每一层 embedding 的维度可能不一致,我们提前做了一些强制操作将 embedding 数据的维度进行对齐。所以针对可解释性,我们可以解释通路,即药物是通过什么路径到达疾病的。但是通路中间 embedding 出来的不同维度所代表的含义是不明确的。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢