

论文题目: Learning Substructure Invariance for Out-of-Distribution Molecular Representations
论文链接: https://openreview.net/pdf?id=2nWUNTnFijm
代码链接: https://github.com/yangnianzu0515/MoleOOD
导读
近年来,分子表示学习(Molecular Representation Learning)获得了广泛关注,目前已有方法已在分子特性预测和靶点识别等任务中均有出色表现。然而,现有方法的模型设计或实验评估过程都是基于训练和测试数据是独立同分布的这样的假设。然而在实际应用中,这样的假设往往并不成立,因为测试分子极有可能来自模型训练阶段未见过的数据分布,这将导致严重的性能下降。
在本工作中,受“不同环境下(例如不同分子骨架、不同分子尺寸等)的分子们的生物化学性质通常与某些分子子结构相关”这样一个现象的启发,我们提出了一个名为 MoleOOD 的新分子表示学习框架,以增强分子表示学习模型对这种分布变化的鲁棒性。具体来说,我们引入了一个环境推理模型,以完全数据驱动的方式识别影响数据生成过程的潜在因素,即环境变量。我们还提出了一个新的学习目标来指导分子编码器识别、编码这些“跨环境性质稳定”的子结构。我们在十个真实数据集上进行实验,实验结果结果表明即使缺少事先人为标注好的环境标签,在各种分布外场景下,我们的模型也具有比现有方法更强的泛化能力。
贡献
预测分子特性对于药物发现和材料设计等许多相关应用相当重要,这类任务通常需要来自化学和药理学等领域的专家付出巨大的努力。近年来,人们提出一系列基于机器学习的分子表示学习方法来加快这些任务的进程,并且在基于机器学习的分子表示学习领域取得了坚实的进展。一般来说,基于机器学习的分子表示学习任务旨在将分子嵌入到隐空间中的向量中,以此表征为基础再用于各种下游任务,例如靶点识别(target identification)、逆合成分析(retrosynthetic analysis)、虚拟筛选(virtual screening)等等。
然而,现有的分子表示学习方法主要基于一个假设:训练和测试的分子数据服从独立同分布(i.i.d.)。但现实世界中的分子数据的分布通常是不确定的,这就需要现有的分子表示学习能够有效地应对分布变化。
本文将介绍一项被 Thirty-sixth Conference on Neural Information Processing Systems (NeurIPS’22) 会议接收的新工作,作者于该工作中提出了一种名为 MoleOOD 的模型以解决上述问题,该工作被选为 Spotlight presentation。
该工作的亮点总结如下:
-
我们首次探索了不变性原理与分子领域知识结合的可能性,并提出了一种能够有效应对分布变化的通用分子表示学习框架叫做 MoleOOD。
-
该框架理论上可以使用任意现有的分子表示学习模型作为骨干模型以提升他们的泛化能力。此外,MoleOOD 能够对训练分子数据自行进行环境划分,并不依赖人为标注的环境标签,所以 MoleOOD也极具实用性。
-
通过在十个公开数据集上的实验,我们提出的 MoleOOD 的有效性也得到了充分验证。
方法
我们的建模动机来源于一个已经被来自生物信息学、药理学、数据挖掘等领域的相关工作提出的观察发现 [5,6,7,8]:分子的生物化学特性通常与一些特殊的分子子结构相关。我们正是基于此先验知识来设计我们的模型。下面我们又给出了两组具体的例子:
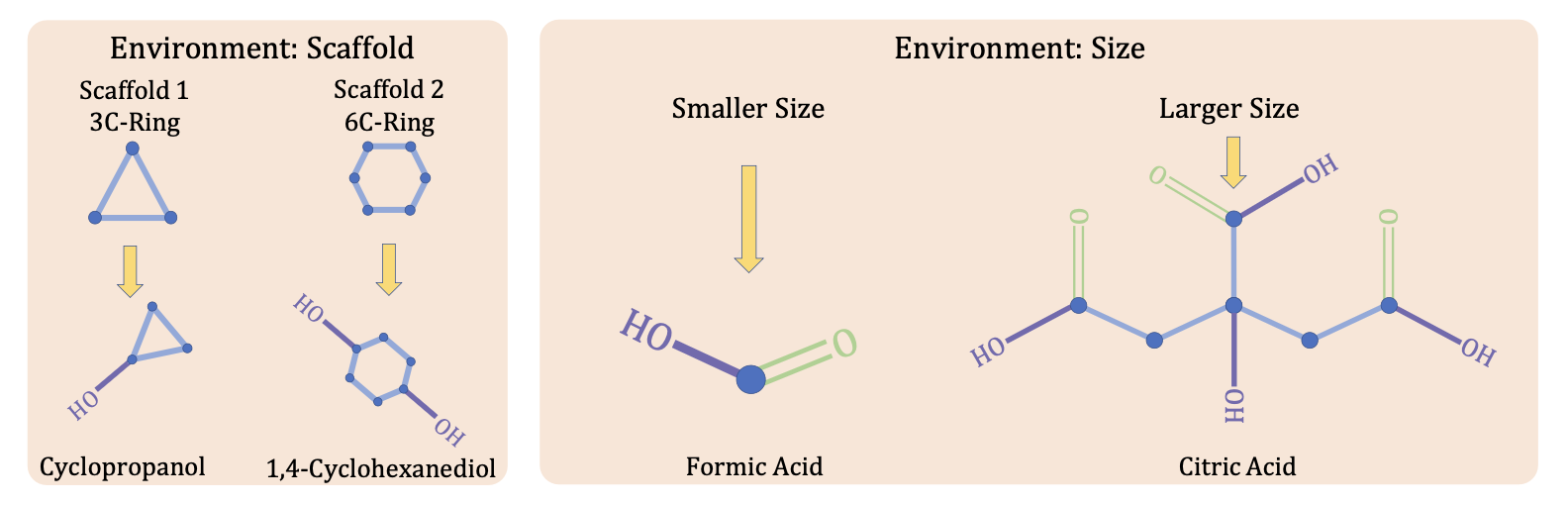
先看左边这一组分子,可以看到他们的分子骨架(scaffold)不同,一个是由3个碳原子构成的环,而另一个是由6个碳原子构成的环。而分子骨架可以被视作一种环境划分的依据,所以这两个分子来自于不同的环境,即来自不同的分布,但他们都含有一个共同的子结构羟基(-OH),所以他们都易溶于水。通过这个例子,我们可以知道羟基和水溶性之间存在一种相对于环境稳定不变的关联性。再看右边的一组分子,可以看到他们的分子尺寸(size)差异明显,分子尺寸可以理解为所含原子数目,而分子尺寸也可以作为环境划分的依据,所以这两个分子也是来自于不同的环境(分布)。
类似地,我们可以发现这两个分子共同含有的子结构羧基(-COOH)也和水溶性之间存在一种相对于环境稳定不变的关联性。我们把分子中这类相对于环境能稳定不变地决定分子性质的子结构叫做不变(invariant)子结构,而分子中其余和分子性质在跨环境时并不不变地稳定相关的则称作虚假(spurious)子结构。因此,一个可行的方法就是从跨环境的和分子性质具有不变关联性的子结构中学习关于某种性质的不变性,从而达到 OoD 泛化的目的。
我们的模型包含了一个分子编码器 用来学习分子的不变表征以及一个预测器 用以做出最后的预测。由于我们无法知道所有可能的环境,即知道,直接求解公式(2)并不现实。因此,我们的目标转为最小化训练数据中已知的不同环境上的risk的期望:
\( minω,ΦEe[Re(Ge,ye)], s.t. y⊥e ∣ Φ(G) (3) \)
对上式中的一些符号做出解释: 而 表示概率独立。我们用 θ 表示 Φ 和 ω中可学习的参数。区别于公式(2),我们加了一个额外的不变性限制 y⊥e ∣ Φ(G)来限制前文提到的虚假关联。
评估因果关系本身具有较高难度,我们可以从信息论的角度重新思考这个问题。回想我们之前所说的,我们希望给定一个分子G, 分子编码器能够利用这些相对于环境不变的子结构学习得到一个分子表征。我们的目标是最大化 Φ(G) 对于 y 的预测能力,这个是可以通过 Φ(G)和 之间的互信息来衡量的。同时,在给定 Φ(G) 之后的 y和 e 概率独立也是可以通过最小化他们之间的互信息实现。为了方便,在下面的部分中,我们用 z 简单表示 G。经过上述从信息论角度的论述,公式(3)现在可以通过解决如下式子从而近似解决:
\( maxω,ΦI(z;y), s.t. minω,ΦI(y;e∣z) (4) \)
如果我们现在再把 和 的输出分别看成分布 qθ(z∣G) 和 qθ(y∣z) ,公式(4)又可以进一步写成:
\( maxqθ(y∣z),qθ(z∣G)I(z;y), s.t. minqθ(y∣z),qθ(z∣G)I(y;e∣z) (5) \)
至此,我们得到了一个更加清晰的优化目标,但看上去可能还是不知道如何具体实现它。在具体说明它的实现前,我们下面先讨论一下有关于环境变量 e 的一些问题。
在实际中,由于给分子标注环境标签非常麻烦,因此在许多情况下人工标注的环境标签并不能获取。当环境标签缺失时,我们可以根据分子骨架(scaffold)直接对分子进行环境划分。但这其实是不合理的,因为最终的总环境数目会很大。以 Open Graph Benchmark 发布的用于分子特性预测任务的数据集HIV为例,如果我们对于 OGB 使用 scaffold 将分子划分到不同的环境中的策略,假设我们直接将每种分子骨架看成一个环境,那么HIV中的 41127 个分子被划分到 19076 个环境中。它的环境数目要远比别的领域的 OoD 数据集的环境数目要大得多。例如 Camelyon17 是一个用于肿瘤检测的 OoD 数据集,它就只将 455954 个组织切片图像划分到了 5 个环境上。尽管有些数据集会提供事先标注好的环境标签,但他们的环境数目还是很大,这对现有的一些 OoD 方法并不友好。所以,我们提出设计一个环境推理(environment-inference)模型将训练集中的分子划分到一些总数相对而言小得多的环境上。我们将划分得到的环境数目设定为一个超参数 k。
我们基于变分推断(Variational Inference)设计了我们的环境推理模型,主要思想就是用一个变分分布 qκ(e∣G,y)去逼近 pτ(e∣G,y)。我们通过公式推导,给出了训练该环境推理模型的目标函数如下:
在我们介绍完我们的环境推理模型之后,然后我们再回到我们推导得到的公式(6),我们给出了如下一个从代码层面角度很容易实现的等价于公式(6)的具体实现如下,而且上述介绍的环境推理模型也结合到了该式子中:
\( Linv(θ;G,τ)=1∣G∣∑(G,y)∈G∣logqθ(y∣G)−Ep(e∣G)[logpτ(y∣G,e)]∣+βEe[1∣Ge∣∑(G,y)∈Ge[−logqθ(y∣G)]] (7) \)
我们在论文中给出了两个定理表明最小化公式(7)里的第一项和第二项分别对应着使得学到的分子表征 z 满足不变性原理的环境不变性(Invariance)和预测充分性(Sufficiency),并在补充材料中给出了定理的证明。
以下是我们提出的 MoleOOD 模型流程图:
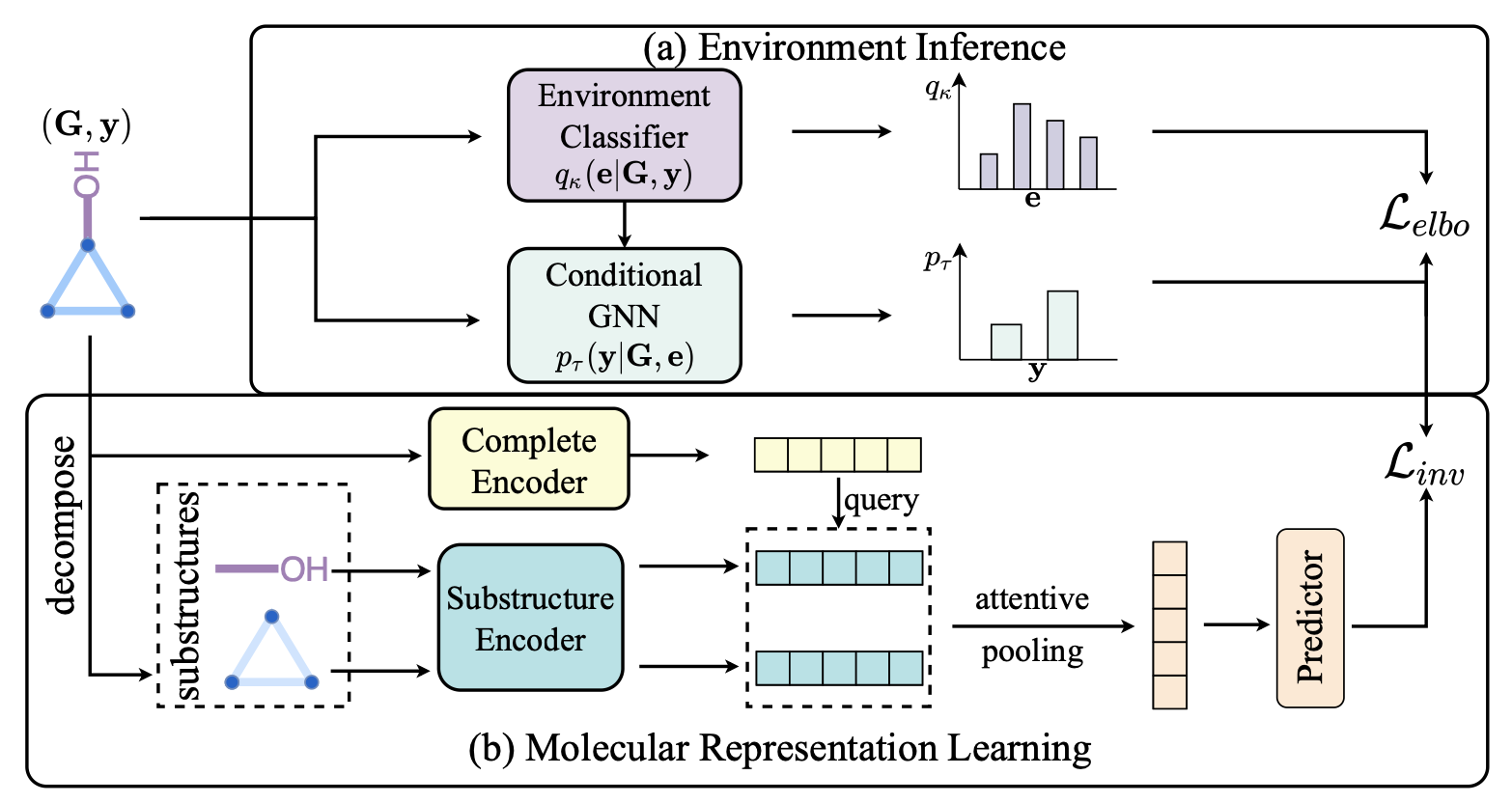
模型训练阶段主要分成两步:
-
我们先训练环境推理模型,这一步是通过公式(6)中的 Lelbo(τ,κ;G) 指导训练过程;
-
然后我们通过公式(7)中的 Linv(θ;G,τ) 指导训练我们的分子编码器和最后的预测器。
实验
我们在Open Graph Benchmark (OGB) [9] 和 DrugOOD Benchmark [10] 提供的共 10 个数据集上都进行了实验来验证我们提出的 MoleOOD 框架的有效性。
数据集: 我们选择了 OGB 中的四个数据集 BACE,BBBP,SIDER 和 HIV,这四个数据集 OGB 是按照分子骨架进行划分的。剩余 6 个数据集来自 DrugOOD,即 IC50/EC50-assay/scaffold/size,DrugOOD 相比于 OGB 提供了更多的划分数据集的方式,即不仅仅可以按照分子骨架(scaffold)定义分布,也可以按照分子测定(assay)或者分子尺寸(size)。所以 DrugOOD 可以提供更全面的 OoD 泛化性能评估。
评价指标: 与之前的分子表示学习相关工作一致,我们采用 ROC-AUC 为评价指标。
Baseline: 理论上,任何现有的分子表示学习模型都可以嵌入到我们的框架中作为我们的骨干模型(backbone)来提升他们原本的泛化能力。我们选取了三种模型:GCN,GIN 和 GraphSAGE 作为骨干模型来验证我们的方法是否会给他们的泛化能力带来提升,并且我们还和他们的增加版模型进行了比较,即加上虚拟节点,也就是下面实验结果表格中的"+ virtual node"。此外,我们还和现有的六种 OoD 泛化方法进行了比较: ERM,IRM, DeepCoral, DANN, MixUp 和 GroupDro。
实验结果: 下面两张表格分别展示了我们提出的 MoleOOD 模型以及 baselines 在 OGB 和 DrugOOD 上的表现。
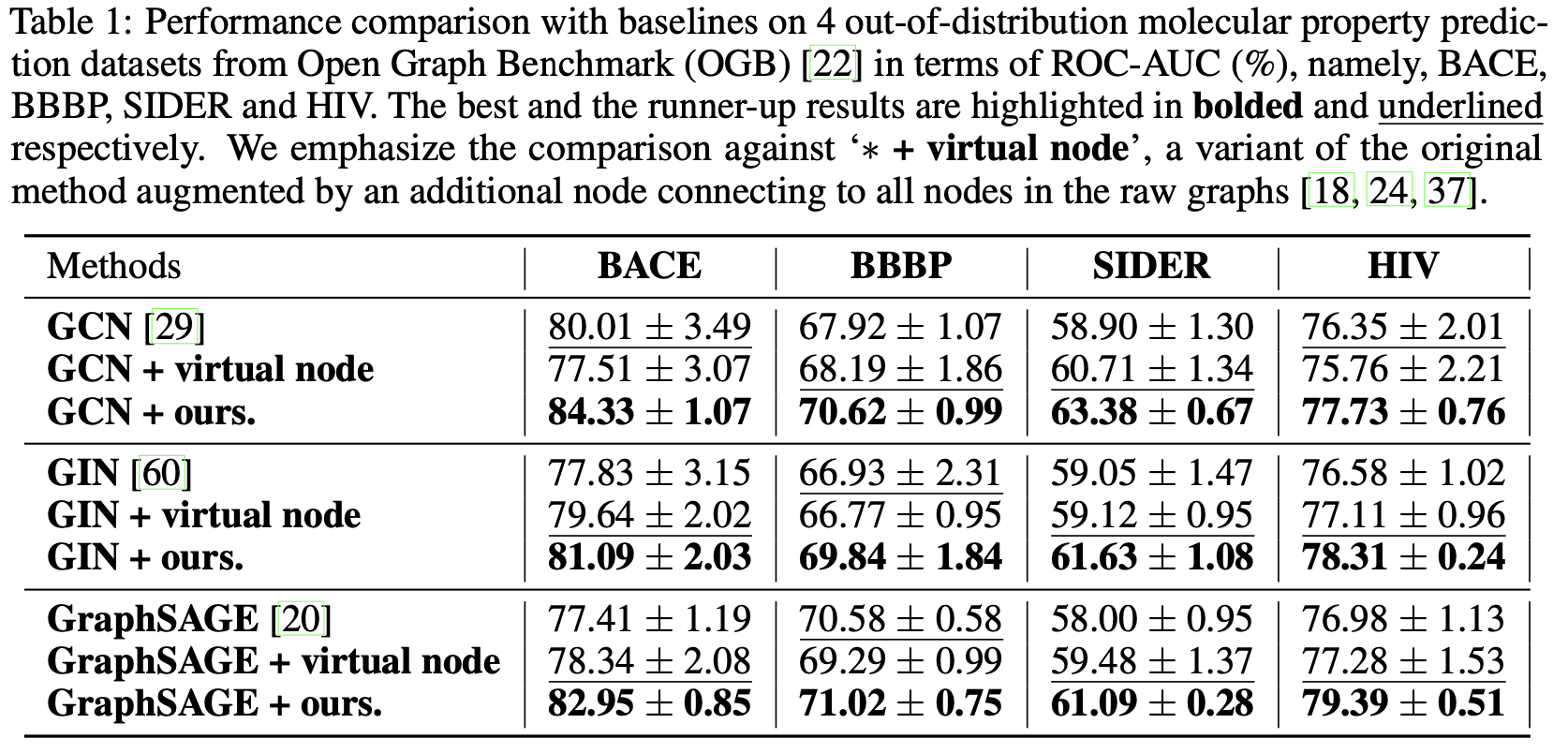
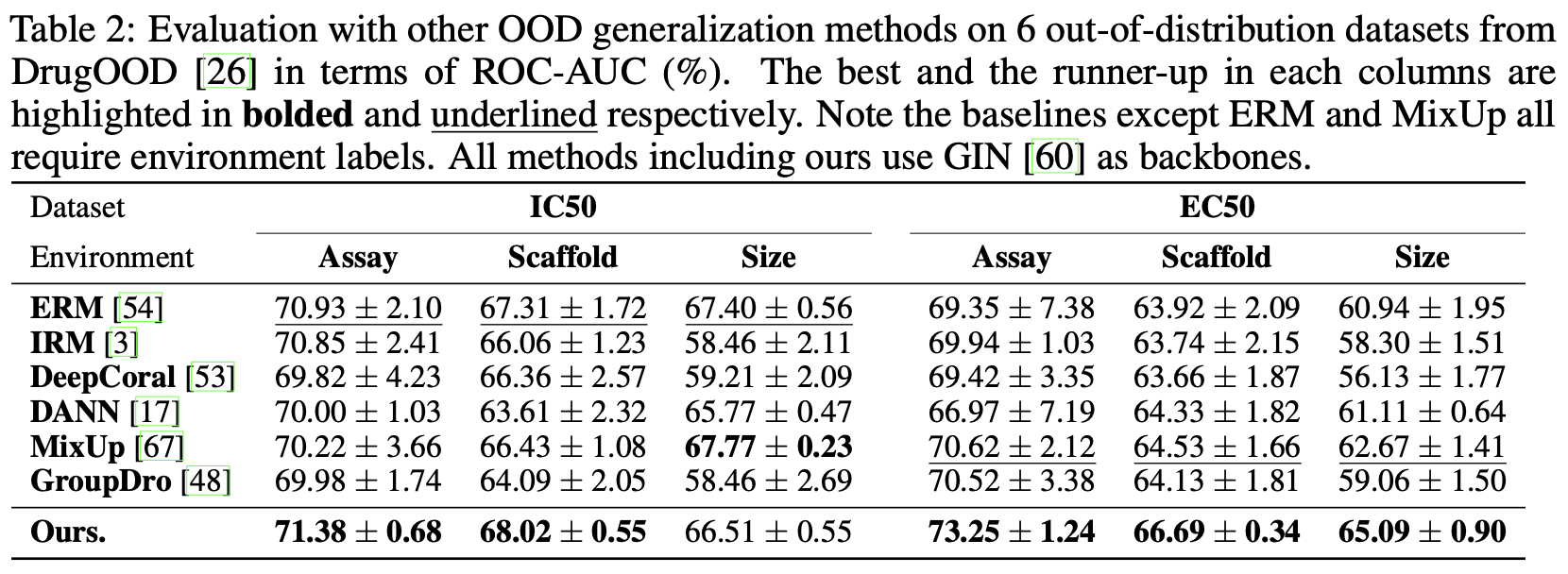
可以看到我们的模型在 OGB 数据集上均给 baselines 带来显著提升,而在 DrugOOD 的六个数据集上,除了 IC50-size 上,MoleOOD 一致地比 baselines 的表现要好。此外,我们还做了一些消融实验来分析模型各个组件对最终表现的影响,比如探究提出的环境推理模型的有效性,这一部分的详细内容可以在论文正文部分看到。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢