
来自今日爱可可的前沿推介
推荐理由:
本文表明AlphaFold,一种蛋白质折叠神经网络,对输入蛋白质序列的小扰动不具有鲁棒性,在对111种COVID-19蛋白质进行评估时表现出显著下降,并强调了对这类网络鲁棒性进一步研究的必要性。
On the Robustness of AlphaFold: A COVID-19 Case Study
I Alkhouri, S Jha, A Beckus, G Atia, A Velasquez...
[University of Central Florida & University of Texas at San Antonio & Air Force Research Laboratory]
AlphaFold鲁棒性:COVID-19案例研究
要点:
-
证明了 AlphaFold,一种蛋白质折叠神经网络,尽管实现了高精度,但不够鲁棒;
-
提出两种度量(RMSD 和 GDT)来衡量蛋白质结构预测的鲁棒性;
-
AlphaFold在对111种COVID-19蛋白质进行评估时表现出显著性能下降。
摘要:
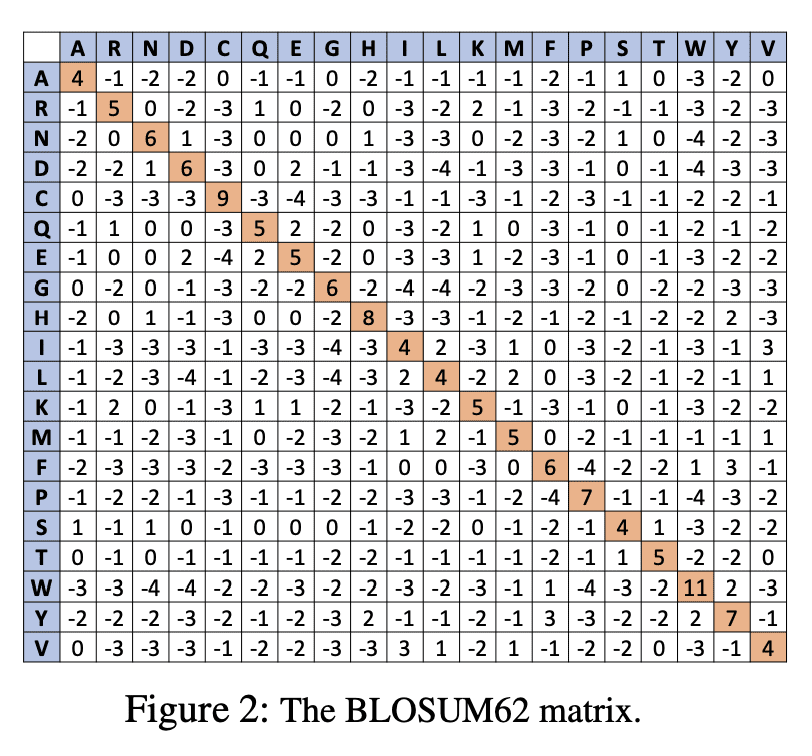
与其他方法相比,AlphaFold等蛋白质折叠神经网络(PFNN)能非常准确地预测蛋白质结构。然而,迄今为止尚未探索此类网络的鲁棒性。鉴于这些技术的广泛社会影响,以及蛋白质序列中生物小的扰动通常不会导致蛋白质结构的急剧变化,这一点尤为重要。本文证明了AlphaFold尽管精度很高,但并不具备足够的鲁棒性。提出了检测和量化这些预测蛋白质结构可信程度的挑战。为了测量预测结构的鲁棒性,用 (i) 均方根偏差(RMSD) 和 (ii) 原始序列的预测结构与其对抗扰动版本结构之间的全局距离测试(GDT)相似性度量。最小扰动蛋白质序列以欺骗蛋白质折叠神经网络的问题是NP完备的。基于成熟的BLOSUM62序列对齐评分矩阵,生成对抗性蛋白质序列,并表明当对抗性变化被 (i) BLOSUM62距离中的20个单位和 (ii) 给定蛋白质序列中的5个残基(数百或数千个残基)所包围时,预测蛋白质结构和原始序列结构之间的RMSD非常大。在实验评估中,考虑了通用蛋白质资源(UniProt)中的111种COVID-19蛋白质,UniProt是由欧洲生物信息学研究所、瑞士生物信息学研究所和美国蛋白质信息资源管理的蛋白质数据的中央资源。这导致总体GDT相似性测试得分平均约为34%,这表明AlphaFold的性能大幅下降。
论文地址:https://arxiv.org/abs/2301.04093
Protein folding neural networks (PFNNs) such as AlphaFold predict remarkably accurate structures of proteins compared to other approaches. However, the robustness of such networks has heretofore not been explored. This is particularly relevant given the broad social implications of such technologies and the fact that biologically small perturbations in the protein sequence do not generally lead to drastic changes in the protein structure. In this paper, we demonstrate that AlphaFold does not exhibit such robustness despite its high accuracy. This raises the challenge of detecting and quantifying the extent to which these predicted protein structures can be trusted. To measure the robustness of the predicted structures, we utilize (i) the root-mean-square deviation (RMSD) and (ii) the Global Distance Test (GDT) similarity measure between the predicted structure of the original sequence and the structure of its adversarially perturbed version. We prove that the problem of minimally perturbing protein sequences to fool protein folding neural networks is NP-complete. Based on the well-established BLOSUM62 sequence alignment scoring matrix, we generate adversarial protein sequences and show that the RMSD between the predicted protein structure and the structure of the original sequence are very large when the adversarial changes are bounded by (i) 20 units in the BLOSUM62 distance, and (ii) five residues (out of hundreds or thousands of residues) in the given protein sequence. In our experimental evaluation, we consider 111 COVID-19 proteins in the Universal Protein resource (UniProt), a central resource for protein data managed by the European Bioinformatics Institute, Swiss Institute of Bioinformatics, and the US Protein Information Resource. These result in an overall GDT similarity test score average of around 34%, demonstrating a substantial drop in the performance of AlphaFold.



内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢