
催化有机反应的机理理解对于设计新催化剂、开发更绿色和可持续的化学过程至关重要,其中动力学分析是从实验数据阐明反应机理的关键。传统的动力学分析依赖于使用初始速率和对数图,并结合速率定律推导。然而,速率定律的推导及其解释需要大量的数学近似,因此很容易出现人为错误,并且仅限于在稳态下只有几个步骤运行的反应网络。近日,来自英国曼彻斯特大学化学系的Jordi Burés和Igor Larrosa团队在Nature 上发表研究论文,通过训练深度神经网络模型来分析动力学数据,可以在无需任何额外的用户输入前提下自动阐明相应的机理类别。该模型以出色的精确度识别各种类型的催化机理,包括涉及催化剂活化和失活步骤等非稳态的机理。
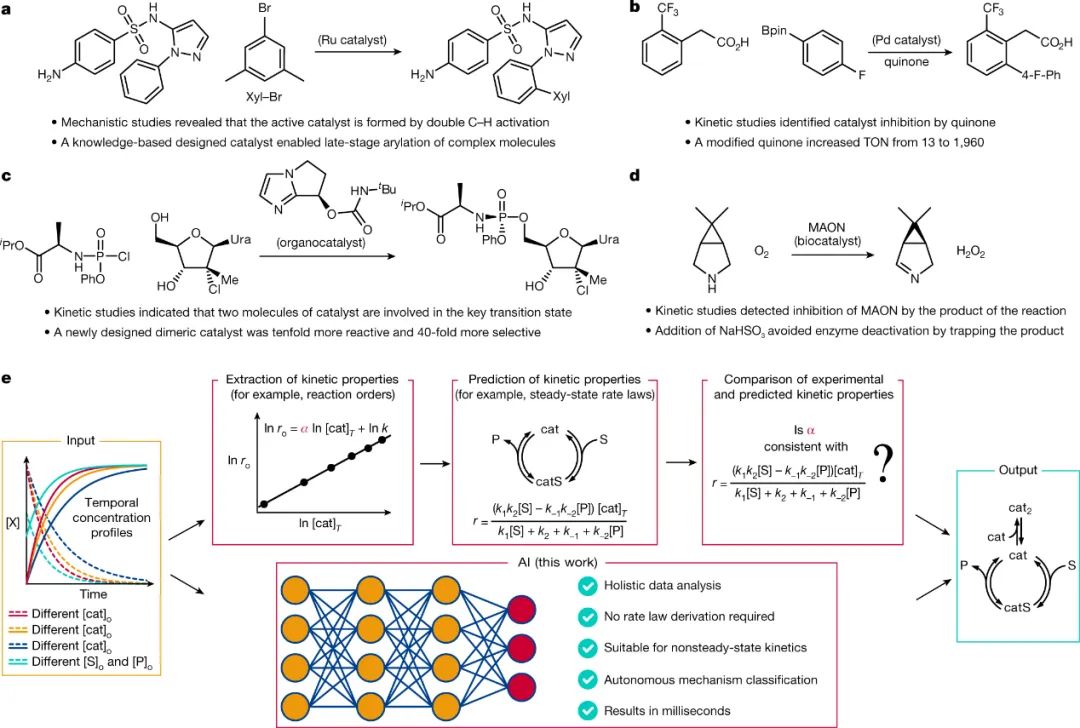
图1. 动力学分析的相关性和最新技术。图片来源:Nature
如图1a-d所示,确定将底物转化为产物所涉基本步骤的顺序,对于合理改进合成方法、设计新催化剂和安全扩大工业过程至关重要。为了阐明反应机理,需要收集多个动力学曲线数据并对其进行动力学分析,尽管动力学数据收集可实现完全自动化,但阐明机理的基础理论框架却未发展到这一水平。当前的动力学分析流程包括三个主要步骤(图1e):1)从实验数据中提取动力学性质,2)预测所有可能机理的动力学性质,3)将实验提取的性质与预测的性质进行比较。尽管稳态方程可以很好地近似许多机理的动力学行为,但无法预测远离稳态的反应系统,例如:催化剂活化缓慢或催化剂不可逆失活的反应。此外,动力学性质的预测还要求化学家充分掌握稳态速率定律的复杂推导和解释。为了简化动力学分析并促进合成实验室对反应机理的阐明,作者基于模拟的动力学数据训练出能够高精度预测来自时间浓度分布的各种反应机理的深度学习模型(图1e)。该模型不需要任何速率定律推导以及动力学性质提取和预测,避免了动力学分析过程中潜在的人为错误,并扩大了可分析的动力学范围,包括非稳态和可逆反应。
如图2a所示,作者定义了20种常见的由催化剂(cat)介导的将底物(S)转化为产物(P)的反应机理。这些机理分别属于四个不同的类别:1)核心机理(M1),这是最简单的 Michaelis-Menten型机理;2)具有双催化步骤的机理(M2–M5),涉及催化剂二聚化(M2和M3)或两种不同催化物种(M4和M5)之间的反应;3)基于核心机理的催化剂活化步骤机理,其中预催化剂需要通过底物配位(M7)或配体解离(M8)进行单分子(M6)活化;4)核心机理的任一催化中间体具有多种催化剂失活步骤的机理(M9-M20),这些机理通常极难区分,但在绝大多数催化过程中都会遇到。
每种机理都由一组动力学常数(k1,... kn)和化学物质浓度的常微分方程(ODEs)函数进行数学描述,这些方程允许生成无限数量的底物和产物的时间浓度曲线,定义一个动力学空间。虽然动力学空间是每种机理的特征,但它们可以部分重叠,例如:如果M2的一组特定动力学常数没有导致非循环二聚体cat2大量形成,那么所得的动力学曲线与M1的动力学曲线难以区分(图2b)。由于不同机理之间的重叠会对模型训练产生不利影响,因此作者使用化学标准来定义每个机理的动力学空间,并优先考虑最简单的机理。
由于深度学习模型的训练通常需要大量数据,因此作者通过数值求解ODEs(无需稳态近似)生成了500万个动力学样本用于模型的训练和验证。用作模型输入的每个动力学样本包含具有固定动力学常数值的特定机理的四个时间浓度分布(图2c),其中三个具有相同的底物初始浓度但不同范围的催化剂浓度;而第四个是“same-excess”实验,包含不同的底物浓度和产物浓度。这种实验组合包含了区分一组潜在反应机理的必要信息,这是因为使用不同浓度的催化剂有助于评估涉及一种以上催化物种的机理步骤,而“same-excess”实验则提供了有关产物抑制以及催化剂活化和失活过程的信息。
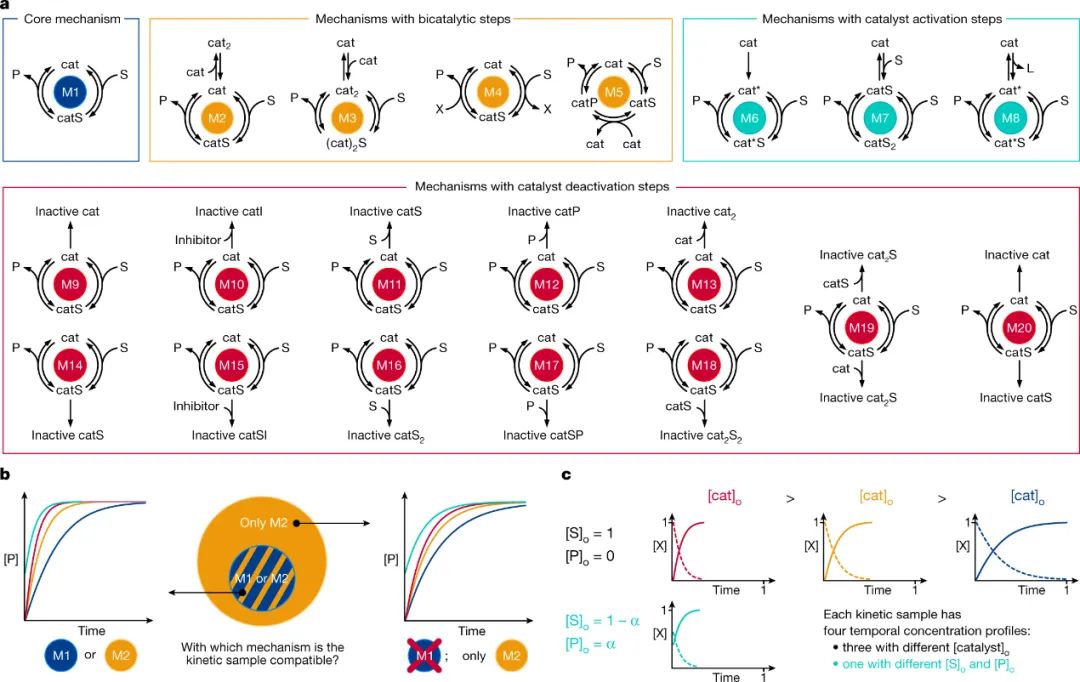
图2. 催化机理的范围和数据的构成。图片来源:Nature
该模型包含 576000 个可训练参数,并结合使用两种类型的神经网络:1)长短期记忆(LSTM)神经网络,一种用于处理时间数据序列(即时间浓度数据)的循环神经网络;2)全连接神经网络,用于处理非时间数据(即每次动力学运行中催化剂的初始浓度和长短期记忆提取的特征),输出为每种机理的概率,概率总和等于1。另外,在训练过程中还进行了数据扩增,包括:1)将样本中的浓度-时间点数量从20减少到20-3范围内的任何值;2)在 S 和 P 的样本浓度值上引入高斯误差。
随后,作者使用100000个动力学样本(每个机理5000个)的测试集评估了训练模型,每个样本包含6个浓度-时间点。值得注意的是,测试集中的所有动力学样本都属于独特的动力学曲线,不同于训练集中使用的动力学曲线。该模型提供了92.6%的分类准确度和100%的top 3准确度,而其它机器学习方法(如:相似性搜索、支持向量机和随机森林)却提供了较差的结果。有趣的是,对实际机理与预测机理的混淆矩阵分析表明大多数机理都被正确预测并具有非常高的召回率(图3a),而大多数错误聚集在2个激活机理(M6和M8)之间和3个失活机理之间 (M11、M12和M14)。
另外,对每种机理的样本预测的概率曲线分析表明在大多数测试样本中,该模型的预测结果非常准确(图3b),而准确度较低的测试样本主要是由于动力学空间的重叠所致。通过允许对预测机理进行分组(图3c),模型分类的准确率高达99.96%(即在100000个样本测试集中只有38个错误),其中69740个测试集样本被预测为独特的机理,23767个样本有2个可能的机理,6067个样本有3个。需要指出的是,该模型倾向于将机理分组在同一类别中,并且类别之间的交叉点很少(图3d),这表明通过单一机理分类获得的最初7.4%的不准确性大部分是由于机理之间的重叠所致。
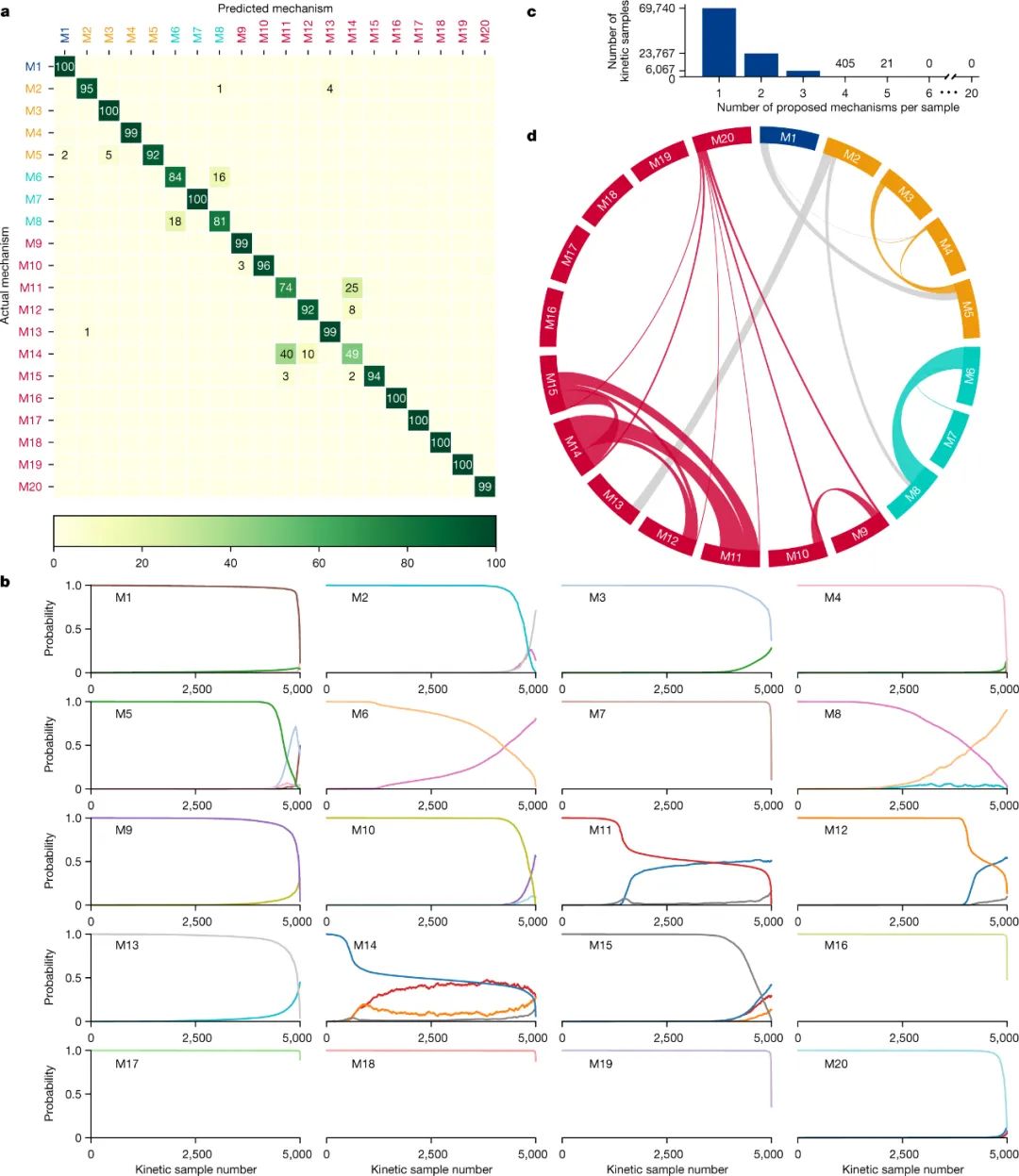
图3. 机器学习模型在测试集上的性能,每个动力学曲线有六个时间点。图片来源:Nature
为了探索模型的潜力,作者研究了引入错误数据和改变数据点数量对模型预测结果的影响,同时在原始测试集的浓度变量上引入了不同级别的高斯误差。如图4b所示,即使测试集数据中存在明显的标准误差(高达2%),该模型仍然能够保持非常高的分类准确度(>99.6%);即使是质量较差的数据(s.e. = 5%),分类准确度也达到了83%。另外,作者还使用1% s.e. 的数据集探索了改变提供给模型预测的浓度-时间点数量的影响,结果显示包含2-20个浓度-时间点的测试集都获得了高分类准确度。该模型能够使用额外的时间点信息来增加正确预测单一机理的比例(图4b,右下角)。相反,当浓度-时间点减少到6个以下时,预测多组机理的数量会增加。
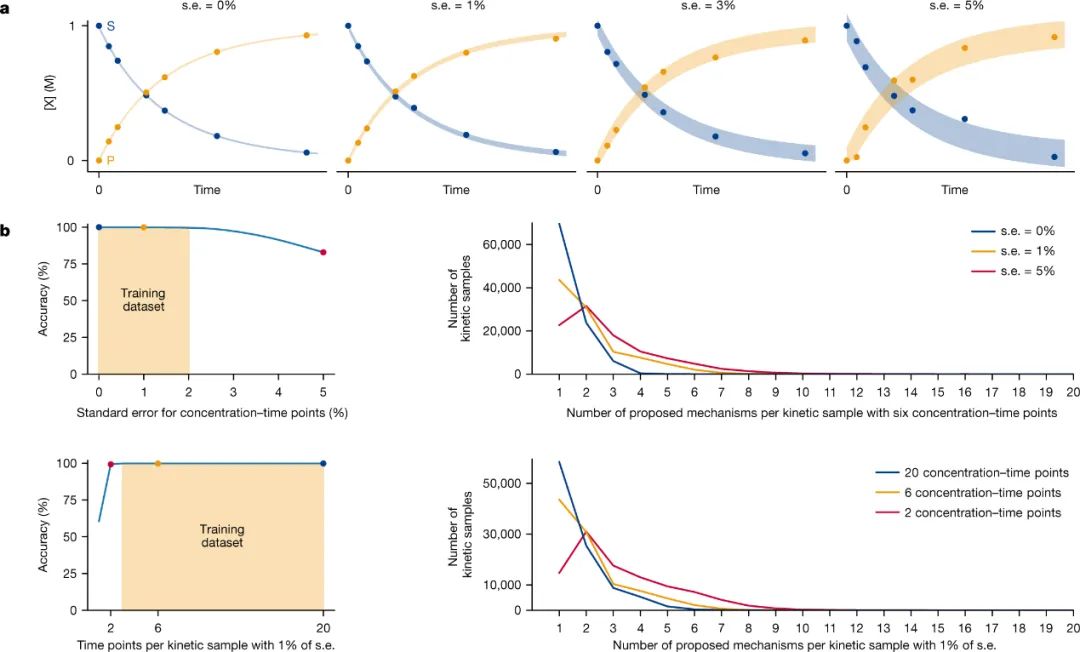
图4. 误差和数据点数量对机器学习模型性能的影响。图片来源:Nature
为了检验人工智能(AI)模型在分析实验动力学数据方面的能力,作者将其应用于各种催化反应中,包括闭环烯烃复分解、环加成、烯烃异构化、C-H键胺化、光催化加氢烷氧化和羰基-烯烃复分解(图5a-f)。图5a-f中每行分别包括:1)具体的反应;2)用作AI模型输入实验动力学数据,其中不同符号分别代表底物浓度(填充符号)、产物浓度(空心符号)、最低催化剂负载量(红色三角形)、中等催化剂负载量(黄色方块)、最大催化剂负载量(蓝色圆圈)等;3)AI模型预测的结果。在所有情况下,AI模型都能够准确识别每个系统的机理特征,并且与文献中通过动力学和其它机理实验提出的特征相匹配。值得注意的是,这些模型还提出了传统动力学分析无法证明的机理,例如:特定的催化剂失活途径。具体而言,Thiel等人报道的闭环烯烃复分解反应(图5a),AI模型不仅能够识别提出的催化剂失活,而且能够将产物牵涉到失活途径,同时该模型的预测结果与先前的DFT计算相一致,表明了乙烯产物诱导催化剂分解的可能性。类似地,Joannou等人报道的[2+2]环加成反应中(图5b),AI模型识别了反应底物参与的催化剂失活。尽管经典的动力学分析由于其对动力学数据的微妙影响而无法识别该机理特征,但在化学计量的有机金属研究中观察到底物介导的催化剂脱氢失活。
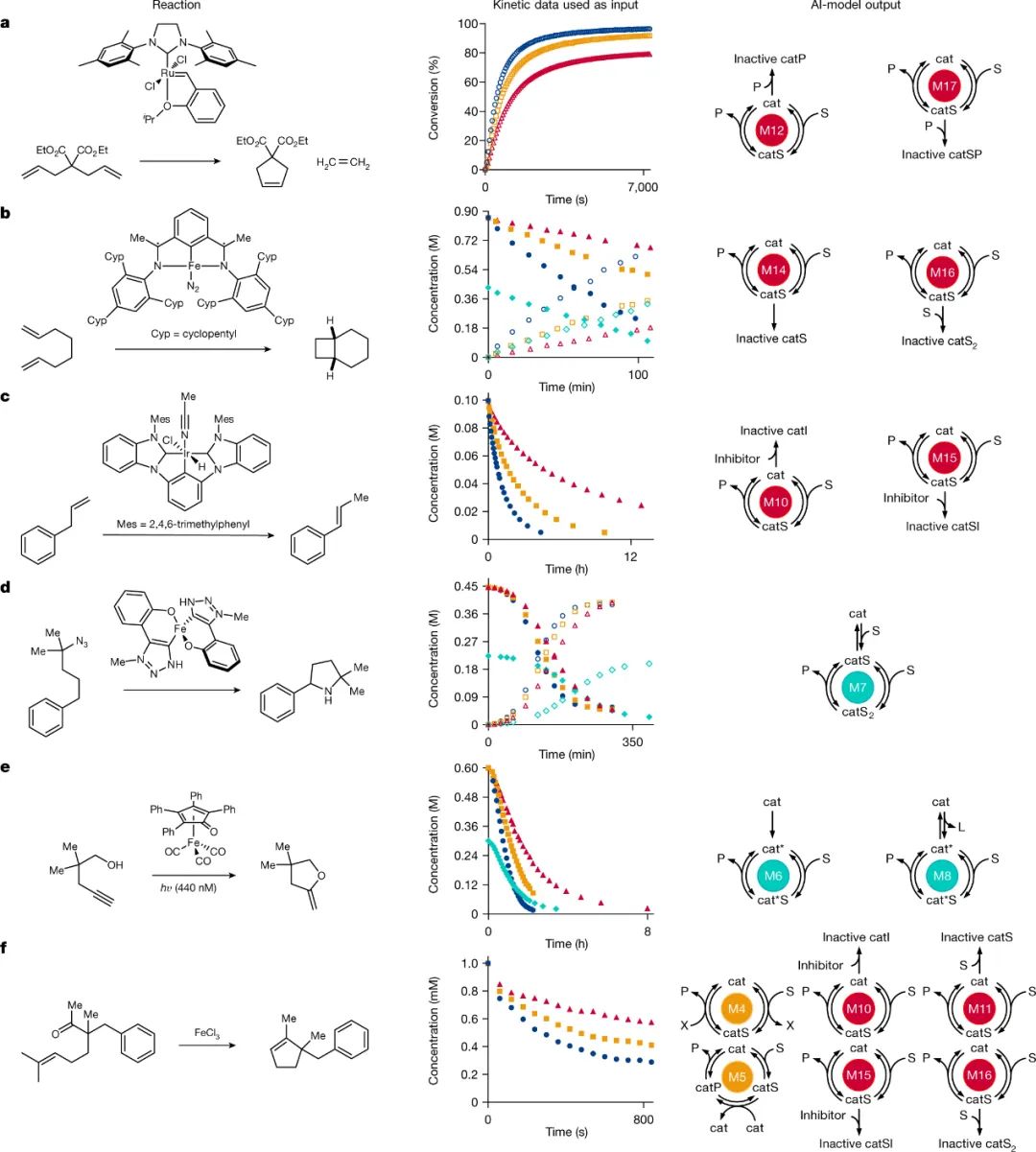
图5. 具有实验动力学数据的案例研究。图片来源:Nature
总结
本文利用深度学习为基于动力学数据的催化机理解释提供了很实用且强大的工具。AI模型将以前冗长的速率定律推导和动力学分析过程简化为一个更方便快捷(只需几毫秒)和准确的流程。经过训练的模型能够解决以前非常具有挑战性的复杂问题,例如:有误差的动力学数据,甚至反应系统处于非稳态的情况。此外,作者还展示了AI模型如何从各种催化反应的动力学实验数据中提取反应机理。
简评
这篇工作本质上是将催化机理问题简化为分类问题(定义了20类反应机理),然后用常规的时序模型(本文用到的AI模型是很常见的)进行分类预测,但所应用的场景和获取数据的方式都让人惊叹。首先,相比于此前AI在化学反应产率预测(Science, 2018, 360, 186-190,点击阅读详细)、反应条件优化(Nature, 2021, 590, 89-96,点击阅读详细)中的应用,AI很少应用到反应机理的解释中。其次也是更重要的一点,不同于前人用的实验(大部分是高通量)数据来训练和验证,本文所用的训练数据是通过解ODEs得到的,就笔者所知,这是AI+化学领域比较罕见的获取数据方式。按照作者的设想,似乎只需要提供适量的动力学分析实验数据,就能以高精度识别正确的反应机理,这是此前AI+化学领域工作中较少看到的实际应用价值。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢