
2023年3月14日,四川大学华西医院华西生物医学大数据中心的陈润生院士团队在signal transduction and targeted therapy发表文章,总结了AlphaFold2的原理和系统架构、成功的原因,以及在生物学和医学领域的应用,此外还讨论了目前AF2预测的局限性。

概要
AF2有望对生物学和医学领域产生重大影响,并可能改变我们进行结构生物学、药物发现、蛋白质设计等相关研究的方式。尽管自AF2开发以来时间很短,但已经有许多与AF2相关的研究报道。为了更好地理解AF2并推广其应用,本文将总结AF2的算法和工作原理及其成功原因,特别是重点回顾其在生物学和医学领域的应用。还将讨论当前AF2预测的局限性。
蛋白质结构预测
到目前为止,已经报道了许多蛋白质结构预测的算法。尽管差异很大,但它们可以大致分为三大类:同源建模、从头建模和基于机器学习的建模。
(1)同源建模
同源建模方法是几十年前最流行的方法。同源性建模的优点包括算法简单、预测速度快以及具有结构已知同系物的蛋白质的高精度。缺陷在于它非常依赖于模板结构,这意味着它无法预测同系物结构中尚未确定的蛋白质的结构。
(2)从头建模
从头建模是一种基于“第一原理”的蛋白质结构预测方法。与同源建模不同,从头建模不依赖于已知的蛋白质结构,而是仅基于既定的物理定律(量子力学)生成目标蛋白质的 3D 结构。简而言之,从头建模方法以氨基酸的原子坐标为变量,在设计的能量函数的指导下进行构象搜索。在这个过程中会产生许多可能的构象,并且选择能量最低的构象。显然,从头建模方法取决于两个因素:(1)表示目标蛋白相对于氨基酸原子坐标的自由能的能量函数;(2)一种有效的构象搜索算法,可以快速识别低能量状态。
关于基于从头建模的蛋白质结构预测有许多研究。从头建模的优点包括:(1)它不依赖于已知的蛋白质结构,这意味着它能够在没有任何先前结构知识的情况下预测蛋白质结构;(2)具有发现新的蛋白质结构类型的可能性。
然而,这种方法面临两个主要障碍。第一个是自由能函数。从理论上讲,自由能的精确计算需要解决薛定谔方程,这需要大量的计算,即使是现在我们也负担不起。因此,必须使用经验公式。目前,大多数经验公式都是基于分子力学或牛顿力学。第二个是蛋白质的构象空间,这是一个天文数字。具有数百个氨基酸的蛋白质的可能构象数量估计约为10的300次方。尽管构象搜索算法以及计算能力和存储空间方面取得了很大进展,但从头建模仍然仅适用于氨基酸残基数在10到80之间的小蛋白质。
(3)基于机器学习的建模
AF2的原理和架构以及AF2成功的原因
AF2是DeepMind最先进的蛋白质结构预测方法。它的原理基于最先进的深度学习算法以及进化中蛋白质结构的守恒。它使用一个新的端到端深度神经网络,该网络被训练为通过利用同源蛋白质和多序列比对的信息从氨基酸序列生成蛋白质结构。
在AF2中,使用了最近开发的一些新的DL算法,其中基于注意力机制的Transformer在提高AF2的性能方面起着关键作用。Transformer是一种新兴的深度神经网络,它应用自我注意机制来获取内在特征,在AI中显示出广泛的应用潜力。
进化中蛋白质结构的守恒是AF2背后的生物学原理。蛋白质在进化中通常是保守的,进化大多是中性的,这意味着大多数突变不会影响蛋白质功能。更重要的是,蛋白质结构比其氨基酸序列更保守。通常,例如,对于远距离物种之间变化80%的序列,3D结构可能几乎保持不变。对齐位置的保持通常意味着它对蛋白质折叠或功能的重要性。蛋白质的两个氨基酸残基的共同进化通常意味着这些氨基酸之间的相互作用。该信息已被用作AF3中2D结构预测的基础。
AF2采用的结构与以前的DL模型完全不同。如图2所示,AF2的流程包括三个模块。
第一个是输入模块
给定一个氨基酸序列,AF2在序列数据库中找到其同系物,并通过比对输入序列及其同系物序列来进行MSA。AF2还检查蛋白质结构数据库中是否有任何同系物具有可用的3D结构,并在氨基酸之间构建成对距离矩阵。然后 AF2 生成 MSA 表示和对表示。应该注意的是,虽然AF2和同源建模都使用MSA,但AF2从MSA中提取并利用协同进化信息,但同源性建模却没有。直观地,当两个残基(A和B)在折叠结构中在空间上彼此靠近时,残基A的突变可能会引起残基B突变的选择性压力。这种共同进化的信息在 MSA 中检测到已被用于辅助 AF2 中的蛋白质结构预测。
值得一提的是,AF2使用了许多高质量的蛋白质序列数据库,包括Uniref90,Uniclust30,MGnify和BFD(Big Fantastic Database)。AF2还利用了几种有效的搜索算法,包括JackHMMER和 HHBlits用于基因搜索,以及 HHSearch用于模板搜索。
第二个是Evoformer模块
Evoformer模块很可能是一个编码器。在本模块中,AF2 从第一个模块获取输入(MSA 表示和对表示),并将它们传递到深度学习模块(称为 Evoformer)。Evoformer 生成经过处理的 MSA 表示和对表示。使用 Evoformer 模块的主要好处是它们能够在 MSA 表示和配对表示之间切换信息:随着成对信息的改进,可以重新解释 MSA 信息,并且以类似的方式,随着 MSA 信息的重新解释,成对信息可以进一步改进。
Evoformer 包含 48 个块,权重不共享。每个块有两个输入:MSA 表示和对表示。每个 Evoformer 模块的输出是更新的 MSA 表示和更新的对表示。MSA 表示和对表示由多个层处理。还使用了 Dropout 方法,该方法通常用于缓解过度拟合的问题。
每个 Evoformer 块(图2b)包含两个基于Transformer的层的路径和两个路径之间的两个“通信通道”。基于Transformer的层的第一个路径作用于MSA。它通过大型蛋白质符号矩阵计算注意力。为了降低计算成本,MSA 注意力被分解为逐行门控自我注意和按列门控自我注意组件。逐行门控自我注意机制允许网络识别哪些氨基酸对更相关,为氨基酸对构建注意力权重。它还结合了来自输入对表示的信息,并且该信息可以被视为一个额外的项。列式门控自我注意允许网络确定哪些序列信息量更大,使属于同一目标氨基酸的组分能够处理信息交换。在逐行门控自我注意和按列门控自我注意步骤之后,MSA 通路具有 MSA 过渡层,其中包括 2 层 MLP。这个技巧增强了注意力机制,并允许它精确定位相互作用的氨基酸对。
基于Transformer的层的第二条途径作用于对表示。该网络的主要特征是注意力是根据残基三角形排列的,这是基于一个直接的原理,即在一个三角形中,任何两条边都可以影响第三条边。这里的直觉是强制执行三角形等价方差。如图2b所示。前两轮更新为三角乘法更新,基于非注意力方法。每个“传出”和“传入”边从包含该边的所有三角形的另外两条边获得更新。后两轮更新是三角形的自我关注。他们更新了 Evoformer 块中的对表示。还涉及两个版本:“起始节点”版本和“结束节点”版本。“起始节点”版本基于具有相同起始节点的所有边更新边。“结束节点”版本的操作方式类似,但它适用于共享相同结束节点的边。成对表示路径还包含三角形自我注意层之后的过渡层,其工作方式与上面介绍的过渡层相同。
第三个是结构模块
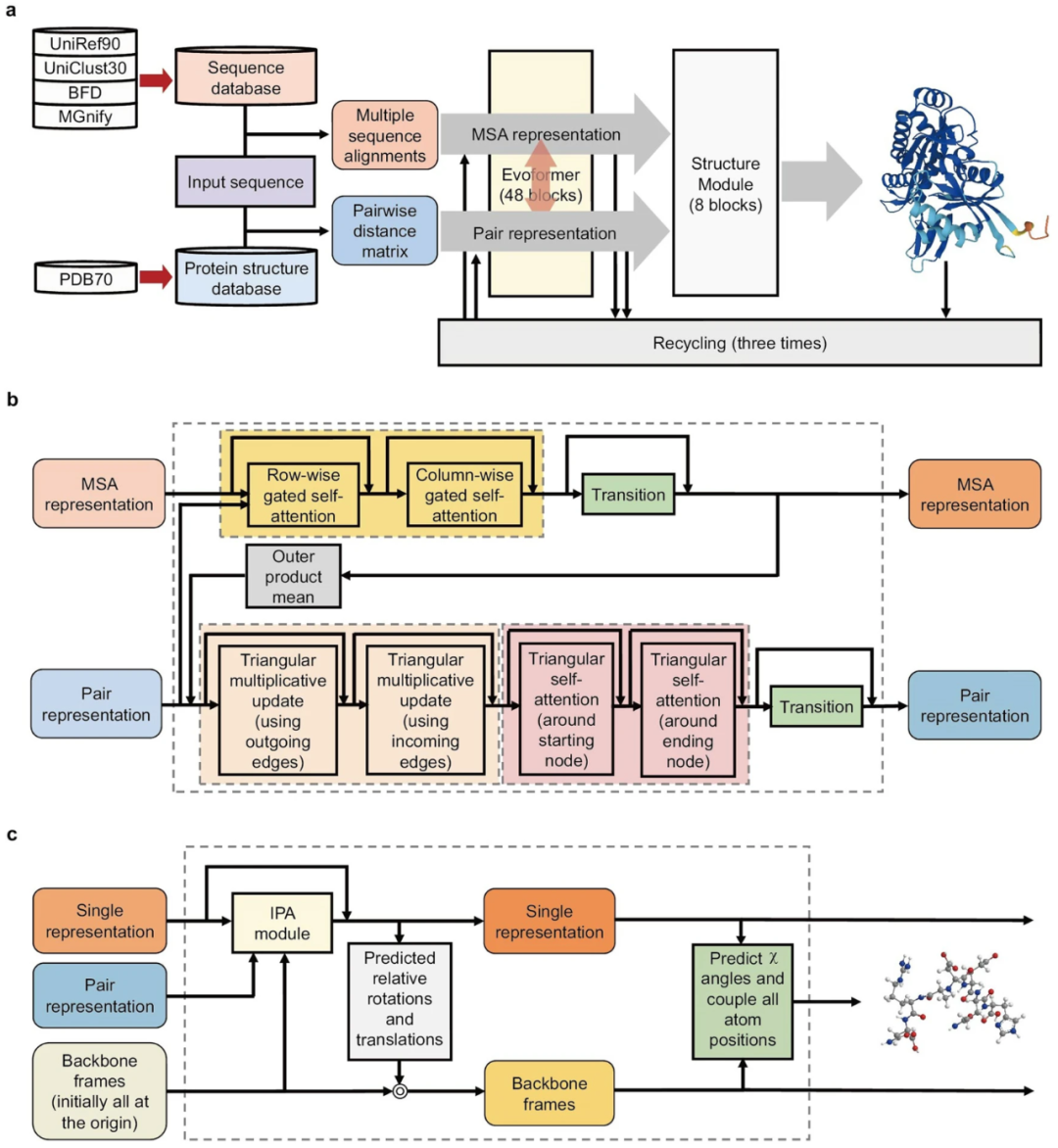
a AF2的总体结构。AF2 的管道包含三个模块。第一个是输入模块,它以氨基酸序列作为输入,并生成MSA表示和对表示。第二个是 Evoformer 模块,它从第一个模块获取 MSA 表示和对表示,并将它们传递到深度学习模块 Evoformer。第三个是结构模块,实现了从蛋白质结构的抽象表示到目标蛋白质的三维原子坐标的过渡。
b Evoformer中一个区块的组成部分。Evoformer 包含 3 个块,权重不共享。MSA 表示形式和对表示形式通过每个块更新。
与以前的版本相比,AF2 实现了最佳性能。虽然我们已经介绍了AF2的原理和架构,但AF2成功的秘诀并没有明确指出。在这里,我们提出了我们对导致AF2成功的最关键点的分析。
从技术角度来看,使用的精细算法是主要原因,这是无可争辩的。其中最重要的是使用基于注意力机制的Transformer。在AF2中,使用了几种类型的注意力机制,每种机制都专注于模型要学习的特定方面。在编码器部分,AF2使用两组相互交织的Transformer:一组主要在原始MSA上运行,另一组主要操作成对信息,通过它们之间的特定信息通道相互更新。MSA逐行门控自我注意允许模型捕获氨基酸序列和蛋白质结构中的长期依赖性。
训练方法也是使AF2成功的一个因素。设计师利用了自我蒸馏的理念。他们使用PDB和预测蛋白质结构的新自蒸馏未标记数据集的组合作为训练AF2的训练数据,其中,25%的训练示例来自PDB中的已知结构,而75%的数据来自新的自我蒸馏数据集。目的是通过使用不同的训练数据增强方法,使AF2回顾以前预测的具有挑战性的蛋白质结构。这种集成数据集方法利用了 AF2 预测的数据,并大大提高了模型的性能。
其他可能有助于AF2成功的算法或技巧包括使用回收方法,从蛋白质数据中学习的端到端框架等。此外,氨基酸序列和结构的大数据也为AF2的成功做出了很大贡献。完整的序列库和足够数量的单域蛋白质结构使深度学习神经网络能够探索蛋白质序列和结构中的各种依赖性,这可能是AF2成功的另一个重要内在原因。
AF2在生物学和医学领域的应用
AF2预测蛋白质结构的优异性能和超过2亿种蛋白质结构的释放正在重塑结构生物学,因此将对需要蛋白质结构信息的生物学和医学领域产生深远影响。AF2及其预测的蛋白质结构将使研究人员有更多机会解决以前被认为极具挑战性的问题。本文回顾了AF2在生物学和医学领域的应用进展。这些应用分为八类:结构生物学、药物发现、蛋白质设计、靶点预测、蛋白质功能预测、蛋白质-蛋白质相互作用、生物学作用机制等(图3)。
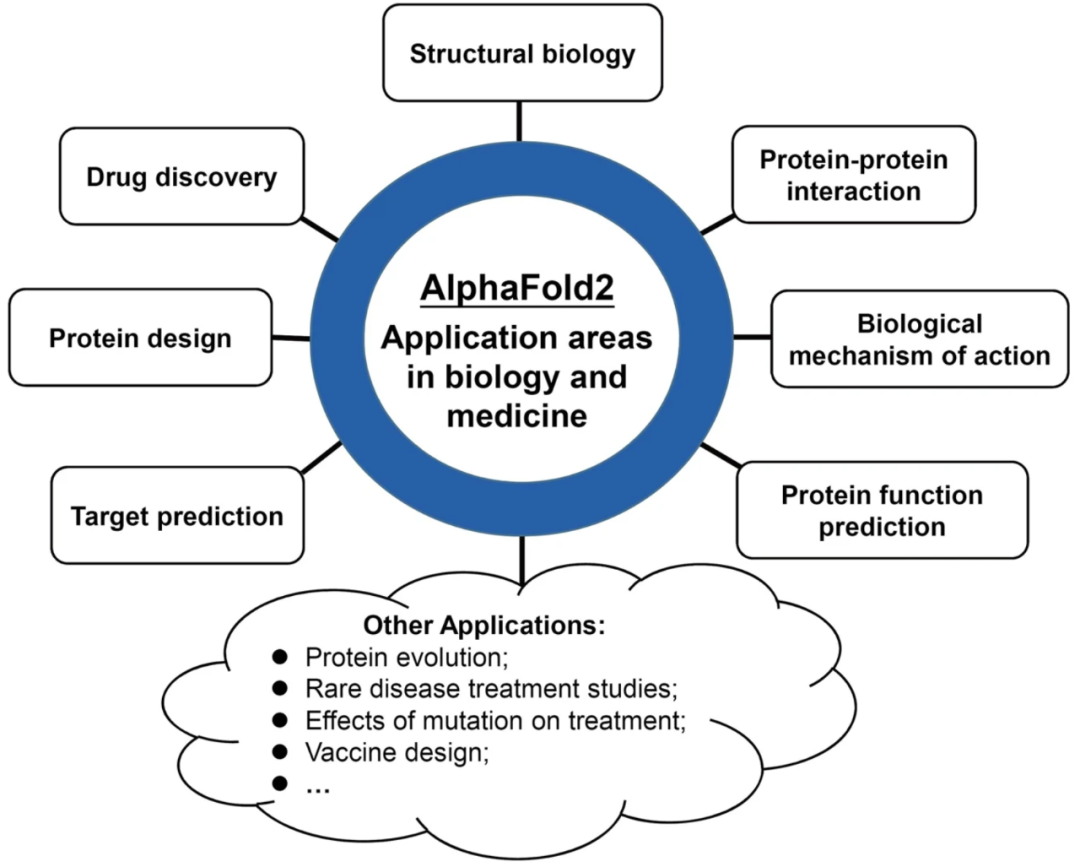
图3 AF2在生物学和医学领域的应用领域
结构生物学
毫无疑问,结构生物学是AF2影响最大的领域。与其说AF2可能会使结构生物学家失业,不如说AF2及其预测的结构将改变我们进行结构生物学的方式。首先,预测结构可以用作求解X射线晶体结构的分子替换模板,这意味着传统的硒代蛋氨酸相几乎没有必要。其次,这些预测的结构也可能有助于通过冷冻电镜确定大蛋白质组装体的结构,冷冻电镜通常需要组分蛋白的结构或其结构域作为拟合冷冻电镜密度的起点。第三,在使用核磁共振求解蛋白质结构时,还可以从预测的结构中受益。通常,使用NMR对结构域或蛋白质进行从头结构测定非常耗时,可能会被AF2结构所取代。因此,AF2预测的应用可以充分利用核磁共振在研究蛋白质折叠和动力学方面的优势。
目前在这方面已经有许多成功的应用。例如,利用 X 射线晶体学和 AF2 预测来确定 B 组轮状病毒中 VP8(一种刺突蛋白)的 VP8* 结构域的结构,利用冷冻电镜和AF2的组合预测揭示了牛痘病毒DNA解旋酶的结构,借助AF27预测、利用冷冻电镜解决了白细胞介素−2信号复合物的结构等等。
还有其他一些类似的研究,其中AF2用于帮助结构测定,甚至可以应用于表达结构的设计。
药物发现
尽管预测的置信水平各不相同,但AF2预测的结构仍然可以大大促进基于结构的药物发现,特别是针对结构信息有限或没有的蛋白质靶点。目前,用于基于结构的药物发现的蛋白质结构主要来自RCSB蛋白质数据库(PDB)。然而,PDB数据库中的蛋白质结构数量相当有限,远远不能满足当前药物发现的旺盛需求。这些结构对整个蛋白质宇宙的释放预计将加速现有和新的药物发现项目。
一些研究表明,AF2建模的侧链质量对于药物发现来说还不够好,最近的一些研究也发现,基于AF2预测结构的对接测试表现出较弱的富集性能。
蛋白质设计
蛋白质的设计意味着创造具有所需结构和功能的新型蛋白质。从头蛋白质设计是合成生物学的长期基本目标。这是一项复杂且具有挑战性的任务,主要受到从氨基酸序列可靠预测蛋白质3D结构的困难的阻碍。AF2 以及其他机器学习算法(如 RoseTTAFold 和最近的语言模型)可能会消除此障碍。可以毫不夸张地说,有了AF2的预测,我们将步入蛋白质设计的新时代。
靶点预测
靶点预测,包括靶向和非靶点识别,不仅对于理解生理和病理过程很重要,而且对于识别新药靶点和评估药物选择性也很重要。靶点鉴定的实验方法,例如各种基于活性的蛋白质分析(ABPP)方法,通常既昂贵又耗时。计算机辅助靶点预测可能有助于缩小靶点识别的范围,靶点识别通常基于蛋白质-配体对接,通常称为逆对接。以前,反向对接面临着缺乏所有可能的蛋白质靶点的3D结构的挑战。AF2结构为开发可行的目标预测方法提供了前所未有的机会。
蛋白质功能预测
目前,仍有许多蛋白质的功能尚不清楚或知之甚少。由于蛋白质的3D结构完全决定了它们的功能,因此可以利用该特性来建立数据驱动的蛋白质功能预测模型。然而,可用蛋白质结构的数量不足严重限制了这些模型的性能。AF2预测的结构为这个问题提供了一个有希望的解决方案,并有望通过增加训练样本的数量来提高这些模型的性能。
蛋白质-蛋白质相互作用
蛋白质-蛋白质相互作用(PPI)是指两个或多个蛋白质分子通过非共价键形成蛋白质复合物的过程。大多数蛋白质需要通过PPI招募其他蛋白质以形成蛋白质复合物以执行其功能。了解相互作用蛋白质的结构是揭示蛋白质功能和机制的基本步骤。然而,缺乏可以产生蛋白质复合物精确结构的计算工具。AF2的出现可以极大地有利于这一领域。
除PPI外,AF2还可用于预测肽-蛋白质相互作用。AF2有望为广泛的肽蛋白复合物提供结构见解。
生物学作用机制
探索生物学作用机制往往很复杂,仍然是一个挑战。生物学作用机制的研究包括药物-靶点相互作用模式、生物酶催化机制等诸多方面。
当前AF2预测的局限性
AF2的发明是结构生物学中改变游戏规则的事件。它通过利用序列信息以原子级精度快速模拟蛋白质折叠,改革了蛋白质结构预测领域。然而,目前的AF2是在蛋白质数据库中的蛋白质结构上进行训练的,其中X射线晶体结构占主导地位。因此,最好将其视为蛋白质可能结晶的实验条件下结构化状态的预测因子,而不是生理条件下最低自由能状态的预测因子。这与方法和技术的一些固有局限性一起限制了AF2预测在许多方面的应用,总结如下。
蛋白质动力学
AF2预测的蛋白质结构是静态状态。然而,蛋白质具有多种状态的动态性。许多重要的生理和病理蛋白(如离子通道蛋白)在不同的活性状态下具有非常微妙的构象变化,并且由于它们与细胞内外的各种其他蛋白质结合,也会表现出不断变化的空间构型。此时,AF2通常给出单一的最优解,难以覆盖蛋白质的构象多样性。然而,这并不意味着不可能用AF2了解蛋白质动力学。根据最近的几项研究, AF2仍可用于蛋白质动力学的一些分析。例如,德尔阿拉莫等人最近提出了一种驱动AF2的方法,用于采样拓扑上多样化的转运蛋白的替代构象以及AF2训练DAT集中不存在的G蛋白偶联受体。
还有其他研究指出,AF2在识别构象歧义方面的表现较弱。此外,AF2几乎不能用于具有多个结构域的蛋白质的结构预测,例如具有大细胞外结构域的跨膜受体。仍然需要设计新的深度学习方法来预测生物物理相关状态的集合。
蛋白质无序区域的结构
AF2数据库包括对大量蛋白质折叠部分的高度准确的预测。然而,在可用于比对的序列较少的情况下,以及天然展开或无序区域的区域,AF2不能很好地预测蛋白质的结构。环结构在晶体中相对稳定,但在溶液中非常灵活。尽管已经尝试了许多方法,现有方法很难预测溶液中蛋白质无序区域的形态、动力学和相互作用。
蛋白质与小分子或其他蛋白质复合物的结构
众所周知,小分子配体或蛋白质可能诱导蛋白质发生构象变化。最具代表性的例子是变构调节剂,其是指小分子或肽与酶蛋白与其内源性配体结合位点不同的位点结合,引起构象变化,从而改变酶的活性。除了变构调节剂外,与内源性配体结合到相同位点的大量正位配体也可以诱导构象变化。然而,AF2并非旨在确定蛋白质在其他相互作用配体或蛋白质存在下如何改变其形状。
具有点突变的蛋白质结构
点突变在蛋白质中经常遇到,特别是病理状态下。了解错义突变对蛋白质结构的影响可能有助于揭示其生物学或病理机制。尽管AF2可以预测野生型(WT)结构,但它在预测错义突变对蛋白质3D结构的影响方面可能表现不佳。尽管有研究表明AF2可以预测错义突变的表型效应,但研究人员观察到AF2错义突变预测的性能并不好,AF2的输出指标与蛋白质稳定性或功能的变化之间只有微弱的相关性或没有相关性。
具有翻译后修饰的蛋白质结构
翻译后修饰,如磷酸化、甲基化、乙酰化作用和糖基化常见于蛋白质中。这些翻译后修饰可能导致蛋白质结构的构象变化。例如,无活性激酶在其活性环中的磷酸化通常会导致较大的构象变化,并最终激活激酶。然而,AF2只能根据其氨基酸序列预测蛋白质结构,并且无法识别残基的翻译后修饰。因此,由于翻译后修饰引起的构象变化无法用当前的AF2预测。
预测孤儿蛋白和人工设计的蛋白
除了上述限制之外,AF2和其他使用DL和MSA中编码的共进化关系信息的计算系统也面临着预测孤儿蛋白和人工设计蛋白的挑战,因为无法产生MSA。最近乔杜里等人开发了一个端到端的深度神经网络模型,即可微递归几何网络(RGN)模型,其中基于Transformer双向编码器表示(BERT)的蛋白质语言模型(AminoBERT)用于从未对齐的蛋白质中学习潜在的结构信息。研究首先引入语言模型从单词中提取语义信息,RGN模型在孤儿蛋白上的性能优于AF2。结果证明了与MSA相比,蛋白质语言模型在结构预测方面的理论和实践优势。
方法和技术的局限性
结束语
尽管自AF2开发以来只有很短的时间,但我们已经见证了许多成功的应用。我们相信,随着时间的推移,人们将会开发出更多的应用或新的应用领域,例如,具有复杂或特定功能的蛋白质机器的设计,新生物体的设计以及疾病诊断。即便如此,AF2预测也不是灵丹妙药,仍有许多问题需要解决,包括蛋白质动力学、蛋白质无序区域的结构、突变体的结构、蛋白质-配体复合物的结构、具有翻译后修饰的蛋白质结构等等。随着AI算法的进一步发展,数据和计算能力的不断增加,预计未来肯定会有更多的惊喜出现。
参考资料
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢