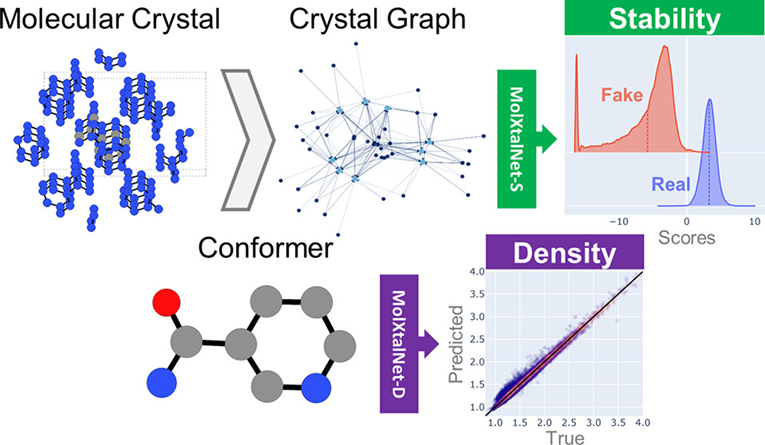
分子晶体的物理性质和生物活性等特征对晶体结构的细节非常敏感,为了确保药物的安全性和有效性或设计出具有所需功能的有机材料,有必要在设计之前确定目标分子可能形成的稳定多晶型体。一般来说,很难实现仅从单分子信息预测晶体结构,因为原子和分子的合理堆积方式数量十分庞大。晶体结构预测(Crystal structure prediction, CSP)通常有两个步骤:(i)搜索可能存在的晶体结构;(ii)对搜索到的晶体结构进行评分,其中评分的方法分为两大类:(i)基于能量的方法,如通用力场(如 GAFF)、特定分子力场和从头计算工具(如密度泛函理论(DFT))等;(ii)基于结构的方法(也称为“拓扑”或“几何”方法)可直接从原子坐标生成得分,而无需对能量进行评估,本文即使用的第二种方法。早在1998年就已经开发了几种基于结构的方法,然而这些先前的方法受到两种关键限制:(i)原子之间的组合数量过于庞大,且不同类型原子对间的距离分布有着显著的差异,(ii) 使用低阶结构相关性,通常为原子的成对距离,会导致模型无法捕获重要的物理特征,而构建高阶结构相关性模型则会导致模型复杂度大大增加。基于以上问题,纽约大学Mark Tuckerman小组开发了基于几何的深度图神经网络(DGNN)的晶体评分模型MolXtalNet-S和晶体密度预测模型MolXtalNet-D,在这些方法中,模型仅需分子晶体中某些片段的特征和分子表面积等信息,而不是直接从原子位置学习几何表示,这大大加快了CSP的速度,同时也拓宽了适用范围,相关成果发表在美国化学会出版的Journal
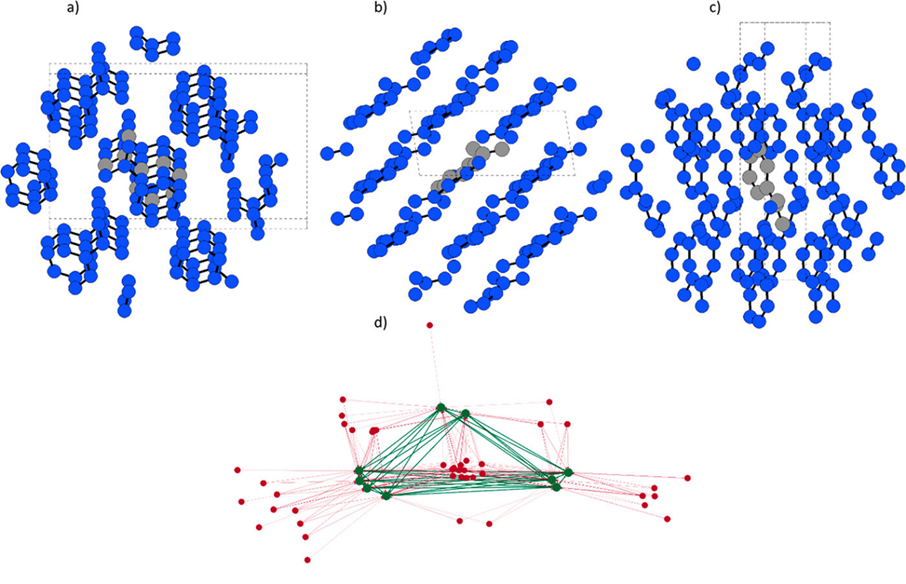
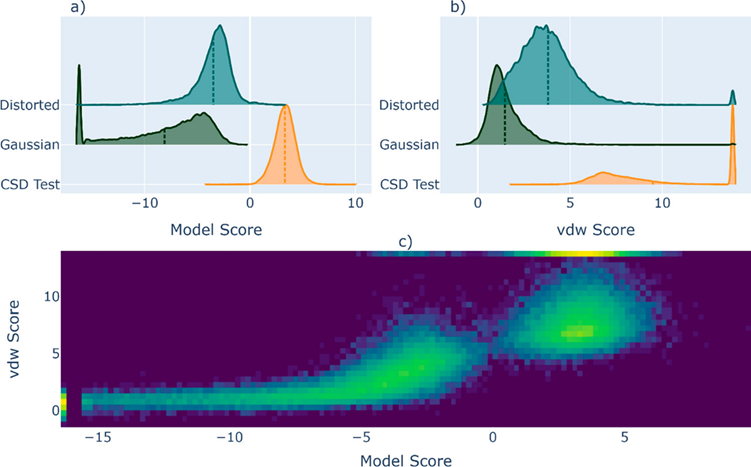
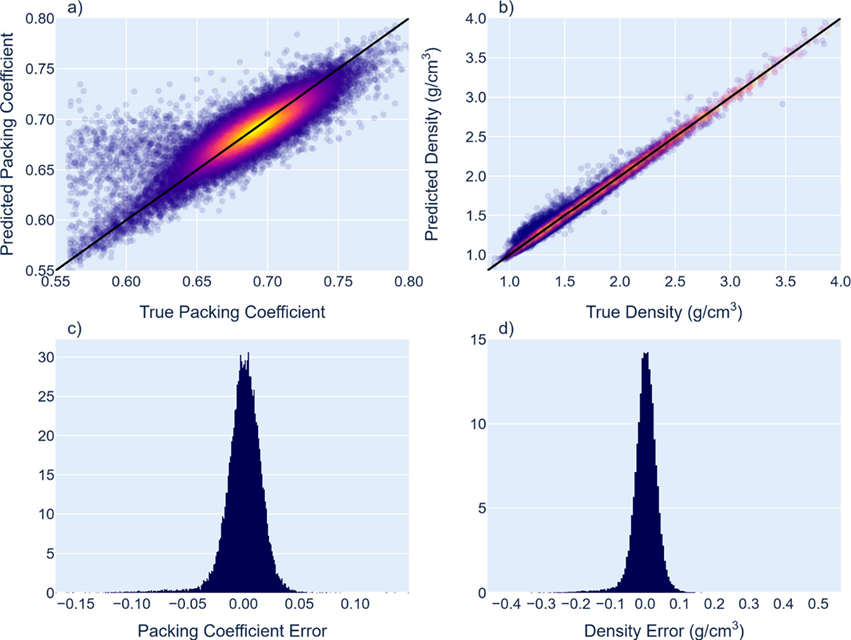
of Chemical Theory and Computation期刊上(J. Chem. Theory Comput. 2023, 19 (14), 4743–4756. )。作者从图像处理领域的填充方法中汲取灵感,开发了分子晶体图卷积(MCGC)方法(图1)。作者首先从剑桥结构数据库(Cambridge Structural Database, CSD)中获取晶体结构,按照80:20的比例划分训练集和测试集,同时限制不对称单元中只含有一个分子的晶体,即 Z′ = 1。接下来对 N × N × N(实际应用中一般为 3 × 3 × 3)超晶胞内的每个原子进行识别和单独标记,具体如下: 0表示分子中处于所选择不对称单元(我们称之为“标准构象”)内的原子;1表示距离标准构象质心 rmax + rc(rmax 是质心与分子中任何原子之间的最大距离,rc是人为设定的截断值)范围内的原子;2表示超出此范围的原子。晶体图由标记为0或1的节点构成,2将被舍弃,同时原子间构建原子标签1→0的有向边。此外,通过将节点特征覆盖到其余对称图像,还可实现对分子晶体对称性和周期性的编码。图 1. 图a) - c)为CSD结构NICOAM03的三视图。灰色分子是标准构象,蓝色分子代表所有参与图卷积的对称构象。图d)为有向图的Kamada Kawai可视化结果。绿色节点对应于标准构象体的原子,而红色节点对应于规范构象体的对称相关图像的原子。绿色和红色连线分别对应于分子内和分子间节点的连接。对于边的嵌入,作者测试了SchNet 、DimeNet 和 SphereNet 几种架构中的嵌入函数,最终选择了最稳定的DimeNet ,使用含有 32 个基函数的贝塞尔公式,省略了角度信息。经过指定层数的图卷积和全连接后,作者并行使用了最大值、总和、平均值和自注意力(SA)等全局聚合器,将结果串联起来并通过一个全连接层,至此产生的特征向量包含模型从分子晶体图中学习到的特征。对于分子晶体的评分,作者使用的是MolXtalNet-S模型。从CSD中获取的晶体结构均为真实样本,此外作者使用了高斯晶体生成器与变形晶体生成器生成假样本,并在相同数量的真实样本和假样本中进行模型的训练。在图2中,作者展示了MolXtalNet-S模型的测试结果,训练和测试中的晶体的畸变程度设置为。图3 a)中显示的分布表明,模型准确筛选出了高斯晶体生成器生成的几乎所有假样本,而变形晶体的平均评分值高于高斯晶体。图 2. a) ,b) CSD 测试数据集(真实样本)和假测试集(高斯晶体与变形晶体)的模型评分和vdW分数分布情况,垂直虚线为分布均值。c) 显示了真实样本和假样本模型评分和vdW分数的二维分布情况。对于分子晶体密度预测,作者选用了晶体堆积系数 作为密度预测指标,并使用MolXtalNet-D模型生成了预测结果(图3)。结果表明,模型对堆积系数和密度的预测值与真实值之间的平均绝对误差仅为1.74%,相关系数分别达到0.853和0.992,回归斜率分别达到0.727和0.982,同样准确地预测了分子晶体的密度。图 3. 堆积系数(无单位,左一列)和密度(g/cm3,右一列)的预测值、真实值回归散点图和误差分布图。黑色对角线对应相关系数为1的完美拟合情况。最后,为了检验模型的通用性,作者对模型性能与分子晶体中各种特征之间的相关性进行了检测。统计结果显示,两个模型与各项分子晶体特征之间都只有较弱的相关性,这说明模型对不同分子的通用性非常好。此外作者还发现MolXtalNet-D模型的损失与堆积系数有较强的负相关关系,表明该模型在致密晶体上具有更优越的性能。本文报道了一种基于几何深度学习方法的分子晶体结构预测模型MolXtalNet,其中包括晶体评分模型MolXtalNet-S和晶体密度预测模型MolXtalNet-D,其核心是采用全分子尺度的特征而不是每个原子的位置信息作为输入。MolXtalNet模型集快速、高质量和广泛的适用性于一身,克服了目前分子晶体结构预测方法耗时且昂贵以及通用性较差的缺点,为分子晶体结构预测提供了有力的新工具。【1】Kilgour, M.; Rogal, J.; Tuckerman, M*. Geometric
Deep Learning for Molecular Crystal Structure Prediction. J. Chem. Theory Comput.2023, 19 (14), 4743–4756.
内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢