
编译 | 曾全晨
审稿 | 王建民
今天为大家介绍的是来自David R. Liu团队的一篇小分子发现的综述。生物活性小分子的开发,作为药物候选物,需要能够访问化学多样性并能快速揭示感兴趣靶点的新配体的发现平台。在过去的15年中,DNA编码文库(DEL)技术已经发展成为广泛使用的小分子发现平台,为许多具有治疗意义的靶点提供了各种各样的生物活性配体。与传统的筛选方法相比,DEL具有许多优势,包括筛选效率高、目标和文库选择容易多重化、评估整个DEL和大型文库所需的资源最小化。综述提供了最近从DEL中发现的小分子的相关报道,包括它们的初始鉴定、优化和生物学性质验证,以及适用于临床应用的合适性。

DNA编码文库(DEL)技术是一种强大的小分子发现方法,首次于1990年代描述,并通过文库合成、选择方法学、数据处理和配体发现的进展而进一步发展。在过去的十年中,DEL技术越来越多地应用于化学探针和新的临床候选物的发现。事实上,自从大约12-15年前首次发现DNA编码的小分子以来,报道新的文库、选择方案和具有生物活性的配体的研究数量迅速增长。DEL技术在学术界和工业界广泛应用,促进了小分子文库的广泛应用。事实上,自2004年的初步研究以来,已经报道了70多个独特的DEL。DEL技术的广泛应用是其易用性、多功能性、高效性和经济性的结果,特别是与传统的基于合成文库筛选的技术相比。高通量筛选(HTS)技术既耗费资源又耗时,通常依赖于需要大量液体处理系统、板操作、分析和储存能力的专用自动化核心设备。HTS需要大量的耗材,包括单独的文库成员、目标物、缓冲液、塑料制品和其他试剂。此外,由于HTS需要逐个成员的分析,这些实验通常需要几天到几周的时间,随着文库的大小和共筛选的目标数量的增加而扩展。需要为每种新的目标类型验证功能性测定进一步延长了总体实验时间。
相比之下,DEL筛选通常可以在几天内完成,仅需要目标蛋白的数十微克,DEL的皮摩尔级别的数量和一套次世代DNA测序(NGS)试剂盒。由于DEL是通过共价连接每个文库成员到一个标识DNA条码(图1a)来创建的,整个文库可以混合在一起并在一个试管中进行目标结合的分析。DEL启用的小分子发现通常使用体外选择来与固定蛋白结合。在将整个混合的DEL与目标蛋白一起孵育后,活性配体将被固定的目标捕获,其共价连接的DNA条码通过PCR扩增和测序以确定与目标结合的配体(图1b)。与HTS筛选相比,这种混合文库选择格式以及DNA标签能够从亚原子级物质中通过PCR扩增的能力,大大减小了筛选的物理规模。与每个新目标类型验证和实施新的功能性筛选测定的需求相比,DEL筛选的缩小规模使得可以同时评估10^5-10^12个成员的文库与许多目标的结合,每个添加目标只需要极少的额外时间和材料。最后,DEL筛选通过NGS对结合的文库成员进行定量读出,无需为每种目标类型验证和实施新的功能性筛选测定。
DEL的实际优势还使研究人员能够通过合同研究机构提供的服务来获取小分子文库。DEL筛选服务现在使得许多学术界和工业界研究人员能够使用强大的小分子发现平台来研究他们感兴趣的目标。然而,其他人选择开发自己的DEL,因为现在有越来越多能力强大和简化的方法可供选择来合成DEL。DEL项目通常能够产生新的具有生物活性的小分子,可用作化学探针或临床候选物,使它们非常适合用于药物发现工作。实际上,几乎所有主要制药公司在某种程度上都使用DEL进行药物发现项目。值得注意的是,迄今为止报道的DEL介导的新型配体中,有一半以上是在过去的6年内报道的(图2a),并且涵盖了各种目标类型(图2b)。
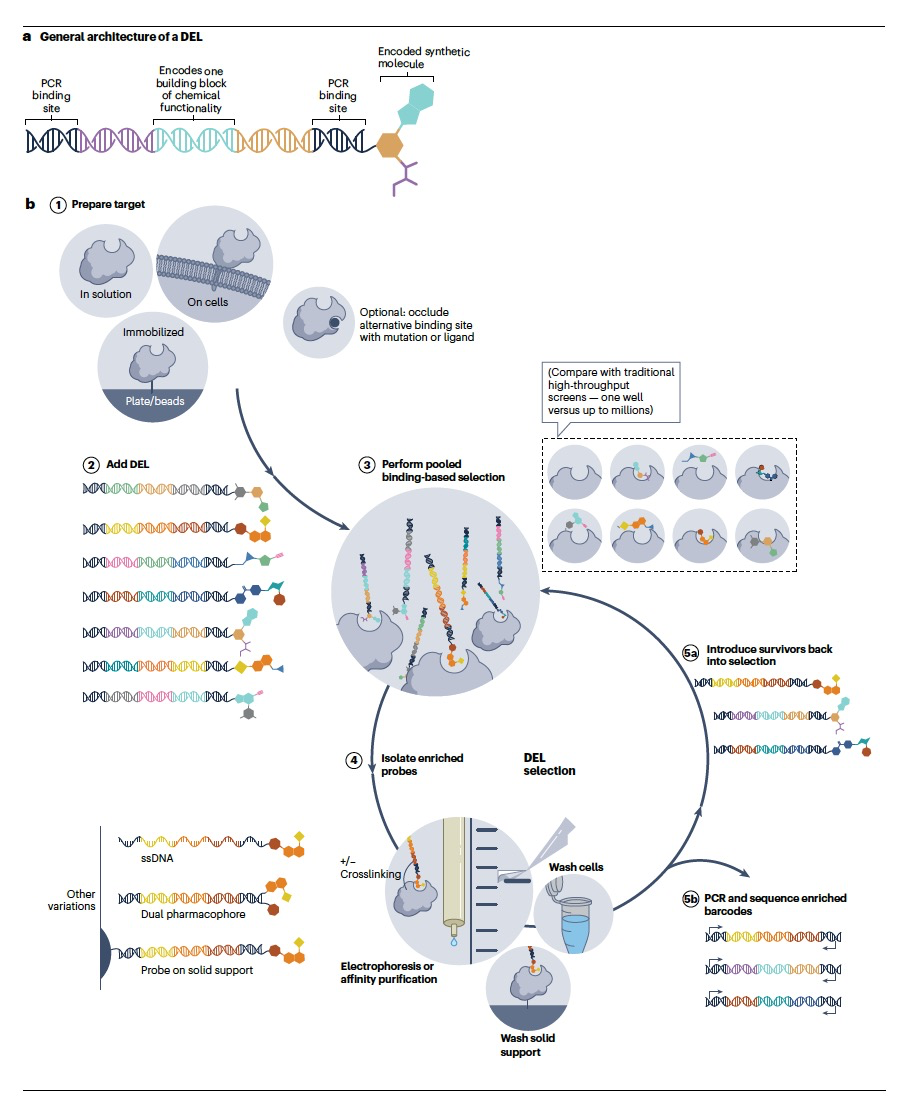
图 1
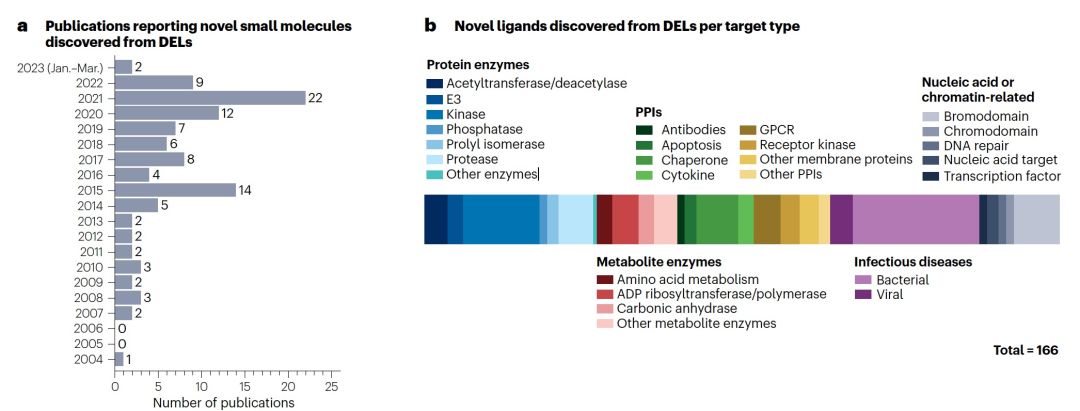
图 2
简单固定化选择中的化合物
应用DEL进行小分子发现通常最常用的是基于结合的单一固定化目标选择(图1b)。这个简单的工作流程可以快速确定与感兴趣目标具有亲和力的新配体,作为优化、表征、进一步开发和临床试验的起点。以下是几个典型的研究,突出了这个过程。在许多情况下,DEL文库也是在同一研究中合成的,突出了文库合成、目标评估和药效学研究之间的方便转化。
与相同规模的非混合库格式相比,混合库格式的DNA编码库(DEL)能够快速合成和应用具有研究者定义特性的新型大型库(数百万到数十亿个成员),加速了新化学物质的发现过程。事实上,在过去几年中,有几个研究团队利用DEL技术快速发现了新的体外探针(表1)。例如,阻止血栓素抑制剂阿加曲班的部分结构被用作支架,合成了一个以蛋白酶为重点的DEL,其成员数量可达980万个。使用固定化的生物素化血栓素进行筛选,发现了一种对血栓蛋白酶活性具有强烈抑制作用的化合物。该研究展示了结构导向的DEL的实用性,并证明DEL能够加速体外新型具有临床相关性的化合物的发现。
在发现新型IL-2结合物的研究中也采用了类似的方法,旨在消除IL-2与IL-2受体α亚单位(IL-2Rα)之间的相互作用,因为α亚单位在免疫抑制性调节T(Treg)细胞中有优先表达的特点。构建了一个包含669,240个成员的DEL(AG-DEL),并与固定化的生物素化L19-IL-2融合蛋白(一种IL-2三期临床试验候选物)进行筛选,因为L19-IL-2比重组IL-2更容易配制和储存。这一筛选过程鉴定出了荧光标记的化合物,经进一步优化得到与IL-2结合强烈且对非靶向血浆伴侣蛋白的结合减弱的化合物。
第三个DEL介导的体外探针发现的例子是针对β-内酰胺酶OXA-48的非机制性抑制剂的发现,它是一种临床上耐药的碳青霉烯酶(图3a)。在构建了一个由三个建筑模块组成的三嗪核心DEL,包含高达1.62亿个库成员后,通过与固定化的His6-OXA-48进行三轮选择,得到了抑制化合物#4,后来缩减为#5。#5与OXA-48结合的共晶结构(PDB ID 6UVK)确实显示其与OXA-48的活性位点和非共价结合。此外,还发现#5可以抑制其他与OXA-48序列同源性成正比的OXA酶。这项研究凸显了DEL介导的新型化合物类型在抗生素发现中的实用性。
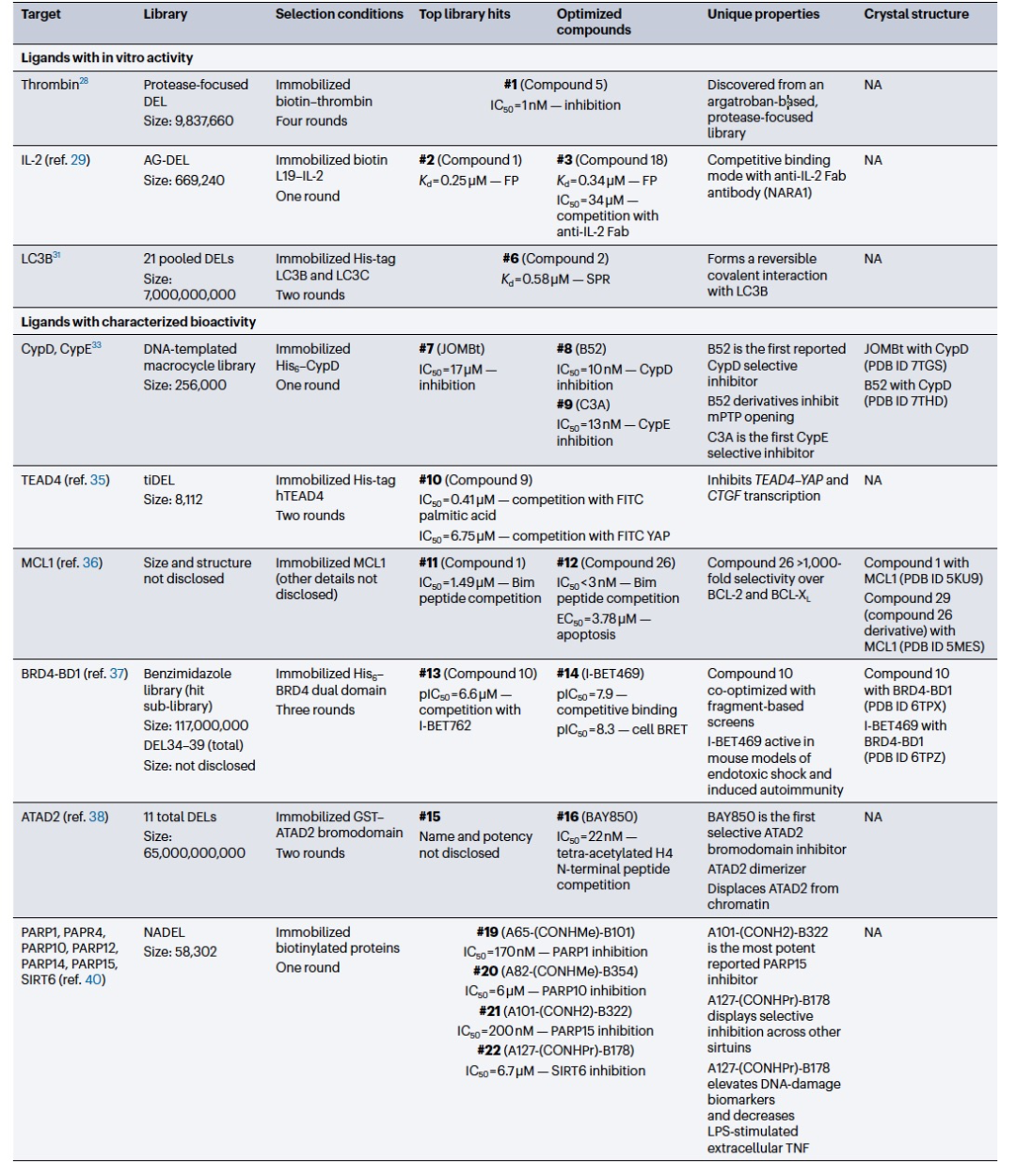
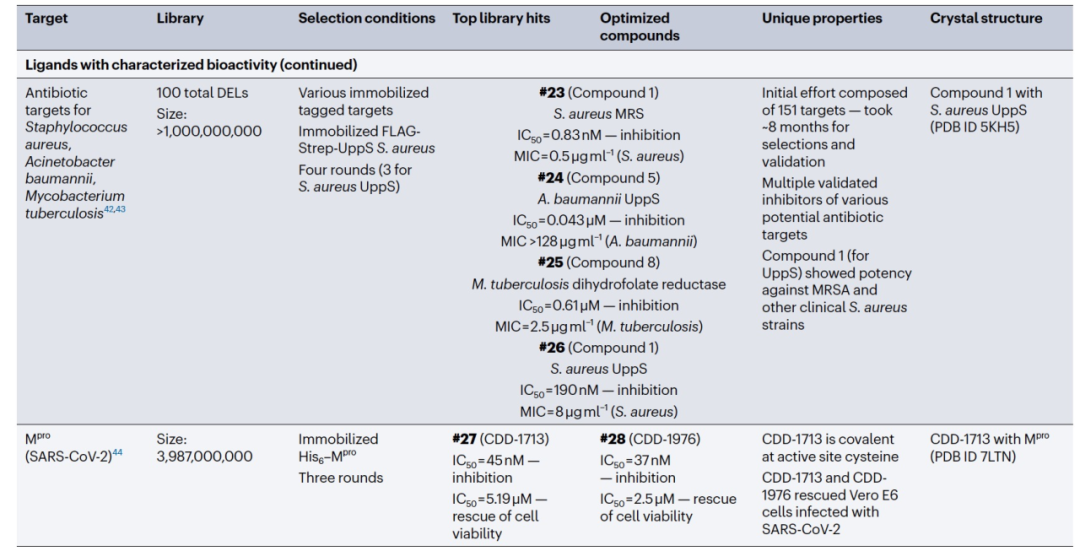
表 1
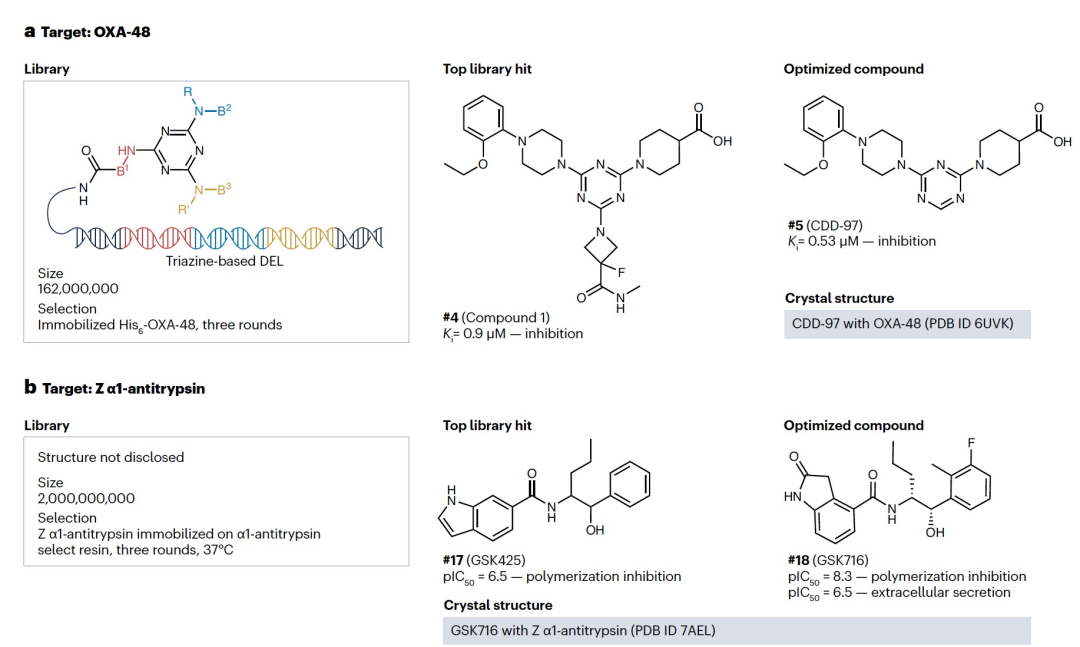
图 3
在过去的6年中,DEL介导了多个靶点类别的活性化合物的发现(表1)。由于DEL选择依赖于基于结合的检测方法,大型DEL可以发现与蛋白结合表面浅的新型配体,这在小分子发现中历来是一个具有挑战性的任务。其中一个明显的例子是对环丙孢酸D(CypD)的新型亚型选择性抑制剂的发现,CypD是一种脯氨酰异构酶,也是线粒体通透性转换孔(mPTP)的关键调节因子。CypD的关键结构特征是两个浅层且暴露于溶剂中的结合口袋(活性口袋和S2口袋)。通过使用最多256,000个成员的宏环DEL进行选择,在固定化的His6-CypD上发现了一种新型活性位点CypD抑制剂(#7),对CypD没有选择性。通过结构引导分析和结构-活性关系,一种二羧酸衍生物#8(PDB ID 7THD)在S2口袋内形成接触,对CypD表现出强效和选择性抑制作用。#8的衍生物同样抑制了孤立线粒体中的CypD-mPTP。为了进一步验证这种选择性结合方式,设计了首个CypE选择性抑制剂(#9),它与CypE的S2口袋K217残基形成可逆共价相互作用。
目前的另一个挑战是蛋白质相互作用(PPI)的广泛但浅层的结合界面。为了应对这一挑战并发现与PPI界面结合的配体,许多研究团队利用了DEL的结合选择。例如,基于色氨酸基序(一种在PPI中富集的氨基酸残基)的tiDEL,其成员数最多达到8,112个,旨在发现翻译增强因子TEF1-4(TEAD1-4)和共转录因子是瘤相关蛋白(YAP)之间的PPI,进而寻找能够消除这种PPI的配体35。通过在固定化的His标记YAP-相互作用结构域上进行选择,PPI抑制剂#10得以发现。由于TEAD-YAP复合物在细胞核中的形成负责CTGF基因的表达,#10显著降低了CTGF转录水平。这项研究表明,即使是具有适当结构基序的中等规模的专注DEL,也可以快速找到传统上难以处理的靶点类别的活性配体。此外,该研究显示,尽管存在DNA标签,DEL仍可支持转录因子配体的发现。
DEL还被用于快速发现凋亡途径中选择性的、活性的PPI抑制剂。利用各种发现策略,包括一系列的DEL,发现了多种癌症类型中过度表达的凋亡抑制剂MCL1的新型抑制剂。DEL平台的选择结果得到了结合化合物#11,它结合到MCL1的BH3结合槽(PDB ID 5KU9)。通过结构优化,得到了大环化合物#12,而与之密切相关的衍生物(PDB ID 5MES)显示出对MCL1的疏水口袋的增强结合。#12在类似的蛋白BCL-2和BCL-XL上的选择性超过1,000倍,并在依赖于MCL1的白血病细胞中诱导凋亡。
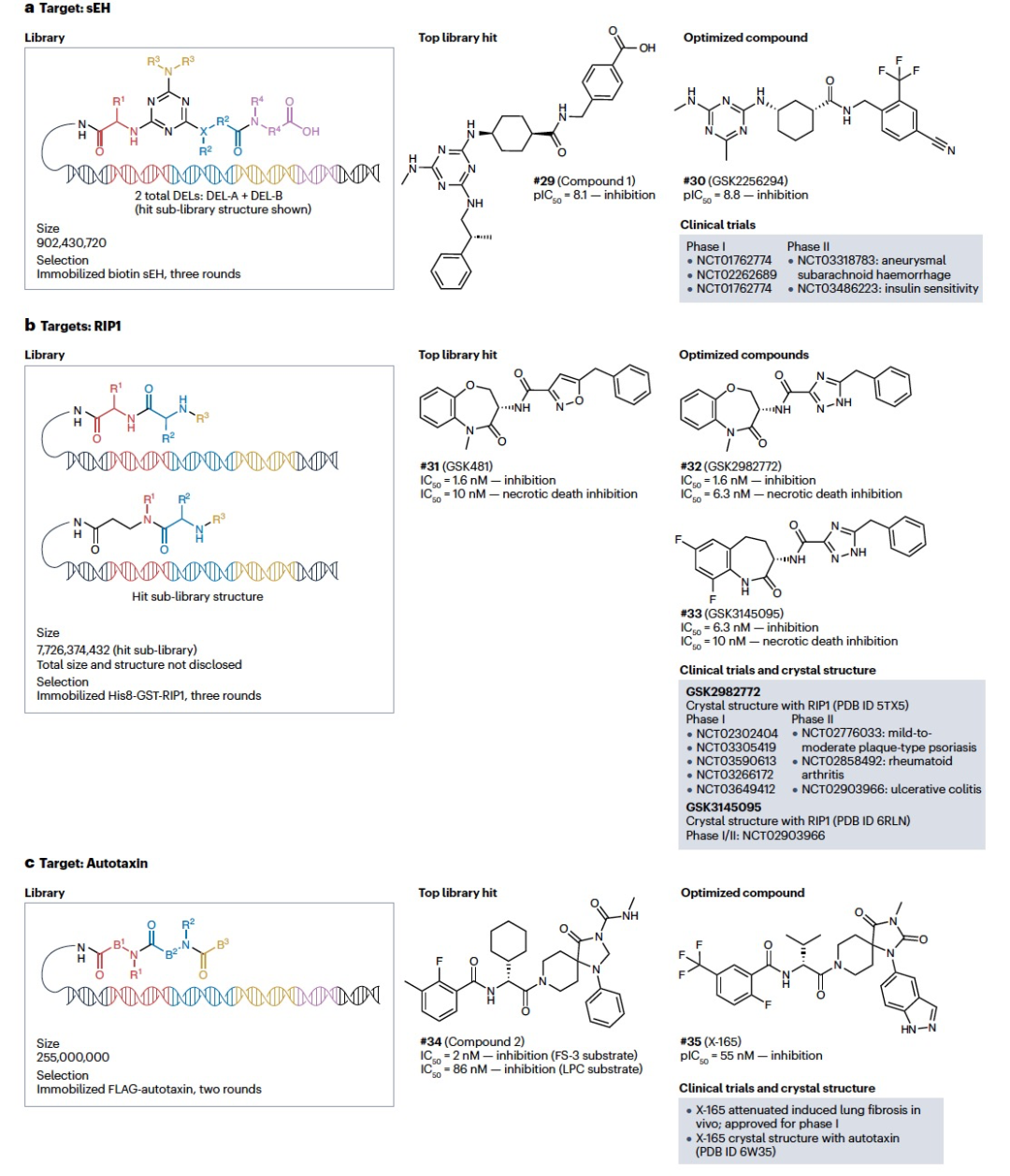
图 4
传统的DEL选择工作流程使用单个固定化蛋白质,已经成功发现了多个临床候选药物。迄今为止,已经对三个临床候选药物进行了评估或批准进行临床试验(图4)。第一个通过DEL实现的临床候选药物是可溶性环氧化酶(sEH)抑制剂(图4a),它是从最初的库筛选结果(#29)优化发展而来的,最终成为了临床候选药物GSK2256294 (#30)。sEH将环氧二十碳三烯酸(EETs)转化为二羟基环氧二十碳三烯酸(DHETs),前者是一种更有效的抗炎代谢物。因此,抑制该酶可能治疗与过度炎症有关的疾病。GSK2256294的安全性和耐受性的I期临床试验已经成功完成(NCT01762774,NCT02262689)。来自2017年的额外I期试验数据(NCT01762774)显示,GSK2256294治疗成功增加了慢性阻塞性肺疾病(COPD)或超重吸烟者患者的EET介导的血管舒张作用和缓激肽介导的血管舒张作用。最近还有两个GSK2256294的II期临床试验的报告:一个用于潜在的动脉瘤性蛛网膜下腔出血的治疗(NCT03318783),另一个用于潜在的治疗肥胖或糖尿病前期患者胰岛素敏感性的改变(NCT03486223)。第一个试验(NCT03318783)报告称,与安慰剂组相比,接受GSK2256294治疗的患者血清中EET与DHET比例大幅增加。在临床终点上,接受GSK2256294治疗的患者住院时间缩短,回家的转归速度比安慰剂组更快,尽管样本规模较小。尽管第二个试验(NCT03486223)仍在进行中,但报道称GSK2256294可以减少患有糖尿病、肥胖或吸烟的患者的氧化应激生物标志物。通过GSK2256294进行的众多临床试验表明,DEL发现平台能够将候选配体转化为临床应用。
工程化DEL选择条件
研究人员通常需要具有特定结合特性的配体,或者对于传统DEL工作流程不适用的靶点需要配体。因此,研究人员尝试了传统DEL固定化工作流程的各种变体。为了解决配体设计问题,研究人员通常利用DEL选择的多重性质,可以将单个或多个靶点与不同的条件共同孵育,以选择用户定义的结合模式。此外,DEL的小规模和易于重新合成的特点使得可以进行额外的选择轮次、优化命中物和改进的结合物。各种研究小组还解决了传统固定化技术不稳定的靶点,并开发了可选择替代固定化或非固定化的体外选择方法。
可以根据需要调整单个选择条件或整个选择过程,利用之前描述的固定化工作流程,以获得具有独特或用户定义属性的配体(表2)。调整条件以增加选择性或多靶点结合物。在最简单的实现中,可以通过依赖并行的工作流程来富集子型选择性配体的命中输出。为了寻找选择性的小分子调节因子,研究人员采用了与相关家族成员进行对比选择的方法。这种方法被用于发现第一个选择性的discoidin结构域受体1 (DDR1)的小分子调节因子,DDR1是一种受体酪氨酸激酶,可以自磷酸化并激活下游的炎症反应。使用两个混合库(分别含有830亿和850亿个成员),对固定化的带有GST标签的DDR1和DDR2进行了选择。通过计算排除同时富集DDR1和DDR2的结合物,鉴定出化合物#36可以选择性地结合到DDR1而不结合DDR2。经过重要的药物化学工作,得到了具有超过1400倍DDR1选择性和超过14倍DDR2选择性的化合物#37,这得益于其独特的激酶结合模式(PDB ID 6FEW,6FEX)。该化合物在Alport综合征的小鼠模型中也显示出治疗效益。这项研究表明,当需要亚型选择性时,可以在主要靶点的同时进行简单的对比选择。
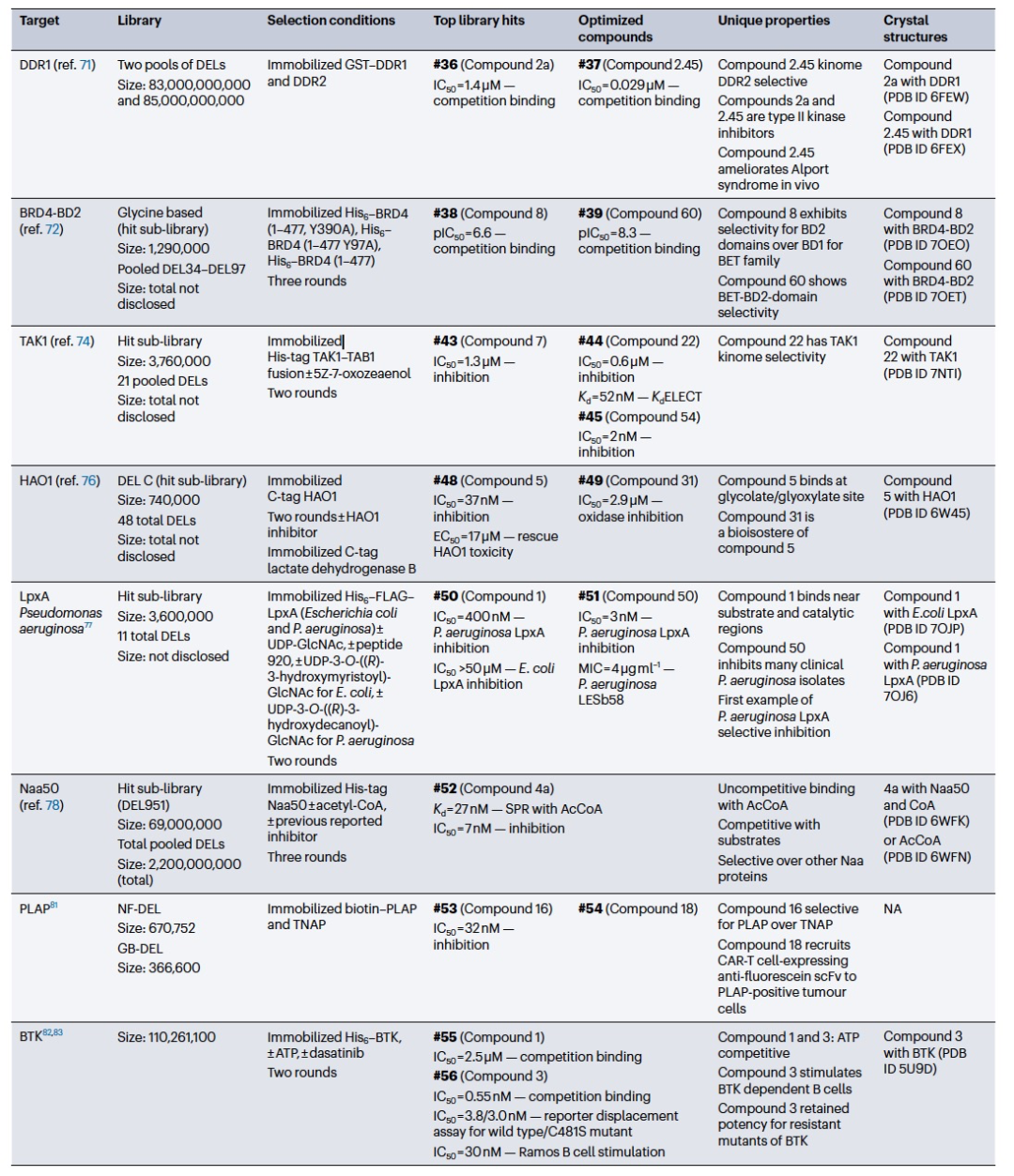
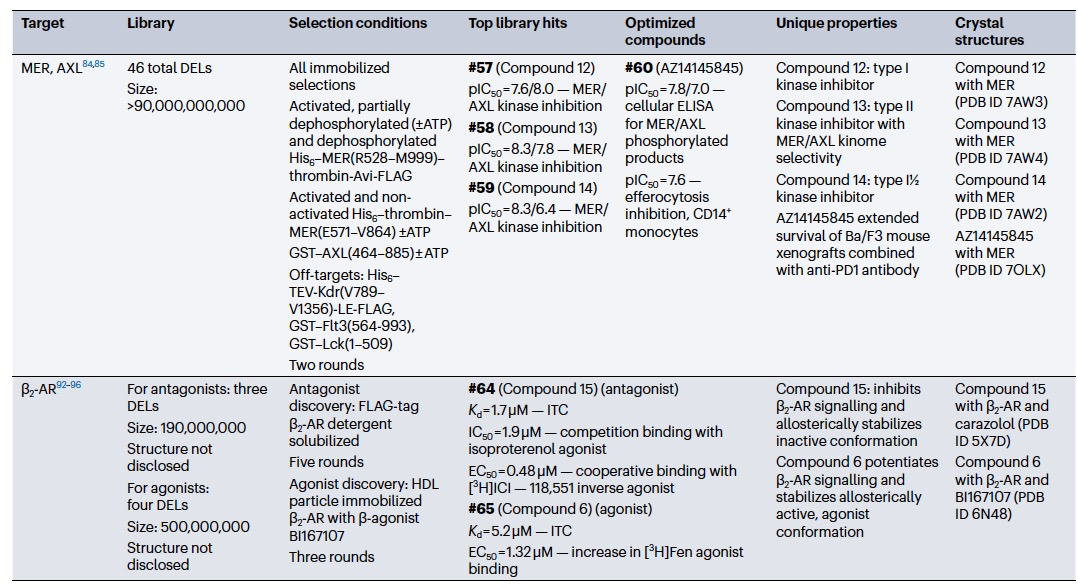
表 2
调整选择输出的方法之一是引入并行选择条件,允许推断库中的某些期望结合模式的配体。为了实现这一目标,可以将库与蛋白质靶点以及具有已知结合模式的其他靶点配体或底物同时孵育。例如,通过引入一个并行条件来确定具有活性位点结合模式的配体,发现了转化生长因子β激活激酶 (TAK1,也称为MAP3K7)的新型抑制剂。使用21个混合DEL,对固定化的His-标记TAK1-TAB1融合蛋白进行了与有或无活性位点抑制剂5Z-7-oxozeaenol共孵育的选择。在不富集于有5Z-7-oxozeaenol条件的子库中,发现了具有潜在活性位点结合模式的抑制剂#43。药物化学和结构引导的优化导致中间体化合物#44和优化化合物#45,表现出对TAK1的激酶选择性。#45与TAK1的共结晶结构(PDB ID 7NTH)显示了一种在铰链区域具有独特的I型结合模式,使得具备激酶选择性,验证了来自DEL选择的与5Z-7-oxozeaenol竞争性结合的观察。这项研究展示了运行并行条件来指导特定配体结合模式的选择输出的实用性。
使用多个DEL选择条件可以覆盖各种可能有用的结合模式,以丰富单个靶点所需的不同类型的配体。例如,由于Bruton酪氨酸激酶(BTK)的抑制剂开始出现临床耐药性,人们寻求新的抑制剂。使用包含高达110,261,100个成员的DEL对固定化的His6-BTK、His6-BTK与饱和量的ATP或His6-BTK与饱和量的达沙替尼(已知的BTK抑制剂)进行了选择。分离出的抑制剂#55在ATP或达沙替尼的选择中富集程度较低。另一个抑制剂#56也在BTK中富集,但在ATP条件下仍保持富集,表明与#55不同的结合模式。进一步的体外机制研究显示,#55与ATP竞争,与选择结果一致。相比之下,#56可能是一种与ATP不竞争的结合物,或者是ATP竞争性结合但非常有效,以至于ATP无法排挤它。#56与BTK的共结晶结构(PDB ID 5U9D)显示了与ATP结合口袋的关键相互作用和一个构象变化,阻塞了进入选择性口袋的通道,表明在ATP结合口袋附近有非常紧密的结合。在后续的研究中,使用不同的体外评估方法,证明了#56能够抑制野生型BTK和临床相关的BTK(C481S)突变体,这是一种半胱氨酸突变体,使之对先前的抑制剂产生了耐药性。这项研究表明,可以使用多个并行的DEL选择来发现一系列具有不同结合特性的配体,以应对单个靶点的需求。
对于膜蛋白和其他难以分离的靶点(如G蛋白偶联受体(GPCR)),DEL选择依赖于其使用体外固定化靶点的优势。然而,这一特性在进行膜蛋白选择和其他难以分离的靶点选择(如G蛋白偶联受体(GPCR))时引发了关注。此外,由于推测的GPCR配体可以具有激动剂或拮抗剂的下游效应,并且体外选择无法选择这些信息,分析变得更加复杂。
在活细胞上的选择
由于蛋白质不稳定性或缺乏成熟的分离方法,一些靶点无法进行任何蛋白质分离或体外选择。此外,与任何基于靶点的结合筛选一样,配体结合质量、靶点的生物化学调节和下游表型之间存在脱节。此外,功能性或基于表型的检测方法(与结合选择不同)已经得到大幅发展和报道。然而,这些检测方法直到最近才被用于发现新的小分子化合物。
在一种创造性的基于DEL的表型筛选示例中,采用了一种固相、颗粒上DEL来筛选针对大肠杆菌和枯草杆菌的新型抗生素。使用了一个多孔颗粒上DEL,包含高达7,488个成员,对细菌琼脂培养基进行筛选。紫外辐射释放颗粒上的活性成分,而活性抗生素在DEL颗粒周围形成生长抑制区域。随后,分离颗粒,通过PCR扩增DNA条形码并进行测序,评估化合物的最小抑菌浓度(MIC)。这些努力导致了新的发现化合物#76的识别。还发现了一些前期报道具有抗生素活性的环丙沙星衍生物。未来的基于表型的筛选可以使用这项技术结合更大的库来探索更大的、新颖的化学空间。
另一项突出DEL的独特应用的研究使用了一个固相肽类DEL,含有高达448,000个成员,用于寻找结核分枝杆菌(M. tuberculosis)感染的非感染性潜伏(LTB)和活动性感染(ATB)状态中循环的IgG的诊断表位替代物。考虑到目前的结核分枝杆菌抗原不能提供适当的特异性或敏感性,宿主免疫状态可能更好地诊断这三种表位替代物。使用荧光活化细胞分选(FACS)进行了一个固相颗粒上DEL的筛选,将其与来自ATB、LTB或非结核分枝杆菌感染个体的血清混合物或单独样本一起孵育。将颗粒与独立的荧光标记的二次抗IgG一起孵育,以标记和区分血清IgG结合的化合物,然后通过重新合成颗粒上的化合物进行验证。使用流式细胞术对血清IgG结合和表位检测进行样本分析,意外发现了一个库的副产物化合物#77与ATB血清结合,对LTB血清的结合有所减弱,并且在非感染血清中几乎没有结合。一个分泌的蛋白质Ag85B,一个二酰甘油酰基转移酶,被确认为#77的抗原模拟物,因为它与#77对血清IgG的结合产生了强竞争作用。
这两项研究颠覆了传统DEL筛选的阶段,传统上依赖于固定的蛋白质和可溶性DEL。固相DEL允许针对更复杂的靶标进行筛选,并在典型的DEL结合筛选之外提供了更多的筛选方法选择。
展望
DEL已成为一种广受欢迎的发现新化学物质的平台。可以调整各种DEL筛选设置以适应目标和所需的配体类型。综述突出了多种在颗粒上、共同孵育或并行筛选条件,帮助研究人员识别甚至选择具有所需属性的配体。尽管大型库(>10^9)可以增强发现工作,但有许多例子表明,聚焦于特定目标的小型DEL已经产生了高度生物活性的小分子。实际上,多种尺寸的DEL中可以包含在当前小分子发现领域中使用的重要化学结构类型。例如,大环化合物可以提供通常与较大生物分子相关的蛋白结合能力,但其生物物理性质更接近小分子,已经开发出了几种大环DEL。共价DEL也已经得到发展,并且现在可以提供一种独特的方法来针对亲核残基,从而增强对关键目标的效力和选择性。基于片段的筛选也已经在DEL结构中得到应用,可以快速筛选出结合片段,然后将其合成为活性配体。由于这些优势,报告DEL介导的新配体发现的研究在过去20年中有所增加(图2a)。迄今为止,DEL所涵盖的生物空间非常广泛,作者预计DEL库及其相应的小分子的使用将继续增加。DEL可以涵盖的目标范围从传统酶到细胞表面受体、蛋白质相互作用和折叠伴侣(图2b)都有所涉及。
未来的目标还包括那些不容易分离或在原生环境之外不稳定的蛋白质。事实上,DEL已经成功应用于血清中、细菌培养板以及针对表面表达的细胞膜蛋白靶标。最近的研究描述了DEL对基于细胞内靶标的适应性,尽管这些努力尚未导致任何成熟的小分子。最后,由于DNA编码库的化学规模远大于其他非基因编码的组合化学库,且选择输出主要是NGS数据,研究人员可以使用各种计算方法来筛选密集的选择数据。机器学习或虚拟组合化学库平台可以根据DEL选择的测序输入预测改进配体的方式,从而实现快速的化合物优化。这项技术在DEL社区内仍然是一个有价值且可扩展的领域。
参考资料
Peterson, A.A., Liu, D.R. Small-molecule discovery through DNA-encoded libraries. Nat Rev Drug Discov (2023).
https://doi.org/10.1038/s41573-023-00713-6
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢