
全文速览
近年来,深度学习在原子系统中的应用迅速增加,包括分子和固态材料。图的使用和消息传递策略的相关设计使得多个深度学习框架能够以比第一原理原子模拟低得多的成本实现对材料性质的可靠和有效的预测。在这篇综述中,我们将重点介绍基于图的深度学习框架的最新发展及其在分子和固态材料系统中的应用。我们将介绍分子和晶体图形表示的发展历史。将回顾由所谓的消息传递定义的基本学习过程,并在此基础上比较不同模型的性能。此外,还将介绍图形学习框架的最新发展,这些框架包含了超出原子水平的材料信息。这一新兴领域处于材料科学、物理、化学和计算机科学的十字路口,本文将给出当前的挑战和未来的展望,重点是如何在图形神经网络设置中组织和组合多层材料信息。
背景介绍
机器学习(ML)技术提供了一种新的机会,通过利用数据驱动的范例来显著降低计算成本并加快材料发现和设计的步伐。与数据驱动技术相结合,ML已成为材料研究的有力工具。浅层ML模型已经被用来预测一大套材料性质,包括相稳定性、晶体结构、电子结构(如带隙)、原子化能以及用于分子动力学模拟的有效势。为了促进分子和固态体系的有效学习,近年来发展了成键原子结构的表示法,如库仑矩阵、键袋和基于多体张量的表示法。
浅显学习模型的一个显着局限性在于,它们严重依赖于基于领域知识设计手工制作的特征。人工智能(AI)的复兴在很大程度上是由深度学习技术推动的特征学习(或表示学习)的突破推动的。这种学习范式使模型参数的自动估计(即端到端学习)成为可能,并将我们从繁琐和非常重要的特征设计过程中解放出来。近年来,为了避免显式特征设计的过程,深度学习被广泛应用于分子和固态材料系统。从原子系统的浅层学习到深度学习,图的概念已经成为综合原子系统和深层神经网络的物质信息收集器和通信器。利用原子之间的成键连接和除此之外的多粒子相互作用,图形是分子和晶体的自然表示。从这个角度来看,量子化学和固态物理正在深度学习范式中融合。基于图形的深度学习框架已经应用于分子和固态晶体材料系统,并显示出与浅层学习相比的前景。
这篇综述将包括深度学习框架的发展,这些框架利用分子和固态晶体系统的基于原子的图形数据。特别是,我们将讨论生成整个输入分子或晶图的函数的多种消息传递算法和聚集过程。我们将介绍图形学习框架的最新发展,这些框架包含了超出原子水平的材料信息。将给出这一新兴领域当前的挑战和未来的前景,以及强调如何在图形神经网络设置中组织和组合多层材料信息。
图解全文
限制传统深度学习技术(如卷积神经网络(CNN))在材料科学中应用的一个关键瓶颈是原子系统中无法用刚性网格描述的巨大多样性。CNN在小分子和正交格子体系的性质预测方面取得了很好的成果。然而,对于非正交网格和大分子,基于CNN的方法被发现是不够的。原子系统中成键结构和多粒子相互作用的存在自然使用图形表示法。原子系统的图形最初是在小分子的训练中引入的。在基于图的学习框架出现之前,大多数ML模型只能处理固定大小的输入。现成的指纹用于计算固定维度的特征向量,作为深度神经网络或其他标准最大似然方法的输入。在D.Duvenaud等人的工作中,计算分子指纹向量的函数被卷积神经网络取代,其输入是代表原始分子的图形。结果表明,从图中提取的有效特征具有更好的可解释性,训练有素的图神经网络(GNN)可以在各种任务上取得优异的预测性能。
接下来,我们将简要介绍ML模型中使用的图的概念。通常,分子或晶体可以用图G来表示,其中单个原子被表示为节点,键被表示为边,或者是有向的,或者是无向的。这些键不一定是化学键,但可以是两个原子之间的任何相互作用,键的选择取决于学习任务。如图1所示,节点作为矢量XV嵌入到图中(对于分子或晶体),而边作为矢量EVW嵌入。可以将可选的全局环境嵌入为向量U。可以重复地将信息交换应用于图形表示,从而在时间步长t处产生更新的状态Xv、Evw和U。
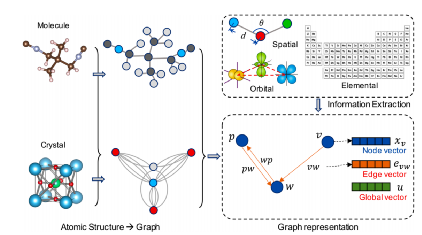
图1 分子或晶体图表示图
基于图形的深度学习的最新发展提供了一种新的工具,当与领域知识相结合时,可以创建分子或晶体结构的创新表示。GNN的一大优势是,它们允许基于任意大小和形状的图形的端到端学习,这有助于消除阻碍深度学习在材料科学中广泛应用的障碍。有了新兴的GNN,任何类型的网格和原子结构都可以在这个框架内成功地建模和分析。此外,分子和凝聚态物质体系的对称性信息可以很容易地集成到图形卷积中。在一般GNN中,卷积运算是可选的而不是强制的,最近的进展为改进复杂材料系统的性质预测带来了另一种可能性。在接下来的几节中,我们将讨论分子和固态晶体系统这两个主要领域中基于图形的深度学习体系的最新发展。
为了有效地捕捉分子内部复杂相互作用所编码的分子结构和性质之间的映射,需要通过设计适当的消息、更新和输出函数来传递和提取分子图中的单个节点和边信息。因此,所谓的消息传递是基于图的深度学习体系结构中的一个基本组件。Google团队的Gilmer等人在几个现有模型的基础上,提出了一个用于图的监督学习的通用框架,称为消息传递神经网络(MPNNS),它抽象了几个现有的图结构数据神经模型之间的共性。MPNN利用了分子系统的对称性,当应用于基于分子的图结构数据时,可以被设计成不同于图的同构。MPNN在具有节点特征XV和边特征EVW的无向图G上操作。节点上的总消息是相邻消息的聚合。然后,通过更新函数更新结点要素。对于T个步骤重复消息传递过程,然后读出函数R计算图形特征。上述工作中的训练集中在拥有134k分子的QM9数据集上。结果表明,具有适当的消息、更新和输出功能的MPNN在4个一般类型的所有13个目标分子性质上实现了最先进的预测结果,并且在13个目标中的11个上实现了化学准确性,表现优于几个强基线,并且不需要复杂的特征工程。
在另一项超越指纹并结合分子图卷积的工作中,作者提出了一种称为编织模的图卷积,它满足以下性质:(i)模型输出不随输入上的排列而变化;(ii)所有时间步长中的原子和对表示(或由该表示计算的值)不随输入不变的排列而变化;(iii)对表示对两个原子的交换不变。经过几次卷积后,使用高斯隶属函数对最终的原子特征进行编码,以获得每个特征维度的模糊直方图。与MPNN不同的是,边状态在卷积过程中被更新,这可以增强信息交换,或者换句话说,节点与节点之间的交互。该模型在三个生化分子数据集PCBA、MUV和Tox21上获得了与基线模型相当的平均曲线下面积(AUC)分数。
2018年,提出了一种连续过滤卷积神经网络(SchNet),通过图形设置来处理分子和固态材料的量子相互作用。SchNet的体系结构如图2所示。这种卷积方案被作者命名为“连续滤光器”。原子特征通过自身与相邻原子之间的相互作用进行迭代更新。这种信息交换过程本质上是传统卷积神经网络在图结构数据上的推广。具有周期边界条件的汇聚层是晶体材料的显著特点,它被应用于网络的末端以产生最终预测。作为第一个可以同时在分子和晶体数据集上执行回归任务的基于图形的深度学习模型,Schnet在包含69,640个大块晶体的数据集上实现了形成能预测的基准结果MAE(0.035 eV/ATOM)。该模型还获得了在QM9数据集上对能量、电子和热物性的预测以及在MD17数据集上预测势能面和力场的基准结果。
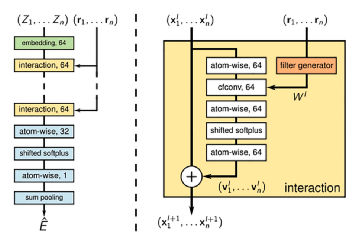
图2 SchNet图解图
第一个将晶体图形与卷积神经网络相结合的工作是由T.Xie和J.C.Grossman于2018年提出的。提出的基于图的学习框架CGCNN将晶体图信息作为卷积神经网络的输入(见图3)。该框架可以从MPNN的总体图中看到,其消息函数为mt(Xv,Xw,Evw)=σ(zi,jwf+bf)⊙g(zi,jws+bs),其中zi,j=Xv⊕Xw⊕evw是中心原子特征向量、邻近原子特征向量和边缘特征向量的级联,σ和g是Sigmoid和Softplus函数。读出函数是全局平均汇集函数,其计算晶体中所有原子的平均特征向量,以获得每个晶体的单个特征向量。该模型当时达到了电子和弹性性质的基准预测精度,如形成能、带隙和泊松比。对于形成能和总能量的预测,MAE分别为0.039 eV/原子和0.072 eV/原子,而对于带隙和费米能量的预测,MAE分别为0.388 eV和0.363 eV。在固体化合物的弹性性能方面,得到了体积弹性系数、剪切弹性系数和泊松比的平均模数分别为0.054、0.087和0.030。模型对金属(非金属)分类的准确率也达到了80%(95%)。
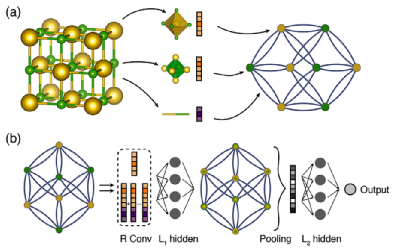
图3 CGCNN体系结构的图解图
在X.Chen等人2019年发表的一项更新的工作中,作者提出了一个通用的基于图的深度学习框架(Megnet),并将其应用于分子和晶体结构(见图4)。在这个新的框架中,在随后的训练步骤中随机地初始化和优化全局特征向量,表示结构的全局状态,如温度或压力。在每个时间步长,图特征由节点、边和全局状态的特征向量组成。新的图形特征是通过卷积函数在它们之间相互作用而产生的。在预测分子轨道能量和热性质方面,该模型优于QM9数据集上的基准模型。对于HOMO/LUMO能级和能隙的回归,获得了0.038 eV、0.031 eV和0.061 eV的MAE。对于不同热物性的预测,该模型优于基准模型。在预测费米能量、带隙、弹性性质和金属-非金属分类方面,它也比CGCNN和SchNet在材料项目晶体数据集上训练得更好。对于费米能和禁带宽度的预测,MAE分别为0.028 eV/原子和0.33 eV。体弹和剪切弹模的MAE分别为0.050 log(Gpa)和0.079 log(Gpa)。该模型对金属(非金属)的分类准确率为78.9%(90.6%)。
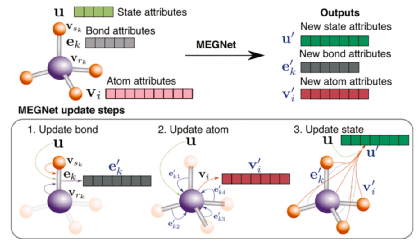
图4 Megnet图解图
作为CGCNN基础上的改进模型,C.W.Park和C.Wolverton提出了所谓的iCGCNN模型,该模型包含了晶体结构的Voronoi镶嵌信息和原子之间的三体关联。此外,还引入了边缘特征的卷积,以使它们在原子卷积过程中保持更新。与CGCNN相比,该模型在形成能量和到凸壳的距离方面要优于CGCNN 20%以上。利用CGCNN和iCGCNN,作者仅通过757DFT计算就筛选了132,600个具有ThCr2Si2晶体结构的元素装饰的化合物,并鉴定了97个新的稳定化合物。这项工作表明,iCGCNN和相关的基于图形的学习框架可以通过快速和准确地识别具有感兴趣的性质的晶体系统来加速新材料的发现。
如上所述,基于原子的图形神经网络已被提出并应用于固体化合物的材料性质预测,与标准神经网络或浅层学习模型相比,表现出相当大的性能改进。虽然取得了很大的性能提升,但仍存在一些根本性的局限性。例如,与能量学和力学性质相比,固态材料的电子结构相关性质被发现更难通过基于原子的GNN来学习和预测。这种基于原子的模型的局限性要求在ML体系结构中融入超越原子的材料信息和物理原理,这是一个巨大的挑战,也是继续发展无机材料人工智能的根本步骤。受鲍林规则的启发,我们最近提出,结构基序,即无机晶体中的积木,可以作为机器学习框架的中心输入。晶体结构中的结构模体是对原子以外的结构信息的更高层次的抽象。这类似于查看分辨率较低的图像,其中一个像素是原始图像中几个小像素的聚合。因此,这些结构基序可以被视为提供更高水平的晶体结构知识的额外指纹。
在我们最近的工作中,研究表明,一种无监督学习算法Atom2Vec可以学习原子的高维向量表示,这些表示编码了原子的基本性质,只需利用材料项目数据库中的化学公式。从原子到基序,我们在最近的一项工作中证明,使用无监督学习算法,可以将大量晶体化合物中结构基序的存在及其联系转换为唯一的矢量表示。我们提出了一个原子基序对偶图网络(AMDNet),它结合了晶体结构信息来增强对与电子结构相关的材料性质的预测。AMDNet由两个不同信息层次的图组成:原子图(其中节点是原子,边是原子间的距离)和Motif图(其中节点是Motif,边是Motif之间的二进制属性,如面角和Motif中心之间的距离)。这两个图都被馈送到如在Megnet模型中使用的图卷积块中。然后将节点和边缘特征传递给set2set模型,以提取与顺序无关的特征。最后,原子和基序特征被连接成总的特征,这些特征被传递到密集的层以得到总体的最终输出(属性预测)。结果表明,在预测复杂金属氧化物的带隙等与电子结构有关的性质方面,基于原子和基于基序的图形的组合优于仅基于原子的图形。
另一层基本的材料信息是原子轨道,它与电子结构和其他性质高度相关。在量子化学领域,最近的两项工作开创了在图中创新性地使用原子轨道来表示分子体系的方法。在2019年发表的SchNOrb(SchNet For Orbitals)深度学习框架中,应用原子轨道的局部基来构建对称适应的成对特征,并将其纳入深度神经网络。SchNOrb的体系结构如图5所示。该模型是先前模型SchNet的扩展,在某种程度上是由SchNet在前层学习原子特征。然后构造成对特征矩阵来表示原子之间的相互作用哈密顿量。在原子轨道特征中采用了旋转对称性。模型输出包括总能量、哈密顿量和重叠矩阵,用于评估组合回归损失。由于显式使用原子轨道表示,SchNOrb能够预测量子力学波函数,由此可以推导出所有其他基态性质。该模型不仅能够很好地预测分子的性质,而且有助于在降低计算成本的同时辅助分子动力学模拟。
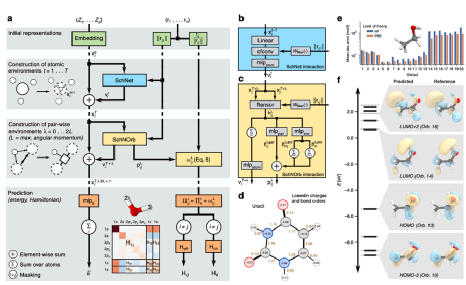
图5 OrbNet图解图
在Z.Qiao等人的较新工作中,作者提出了一个称为OrbNet的模型,该模型以对称适应的原子轨道(来自低成本第一原理计算)为输入,并利用MPNN作为卷积模块(见图6)。消息函数为M(Xv,Xw,evw)=σ(W[Xv⊙Xw⊙evw]+b),更新函数是Ut=Xv+σ(W[⊕i∑w∈N(V)αvwmvw]+b),其中σ是Sigmoid函数,⊙是元素乘积,⊕是级联,W是加权矩阵,b是偏置向量。通过计算每对节点的权重来使用注意机制:αvw=σ(∑(Wxv)⊙∑(Wxw)⊙evw/ne)其中Ne是边缘特征向量的维度。在几次卷积之后,使用读出函数R=∑v∑txv来聚集来自所有时间步长和所有节点的学习特征。OrbNet在学习效率和可转移性方面优于许多以前的ML方法,用于以非常低的计算成本预测分子性质,如能量。
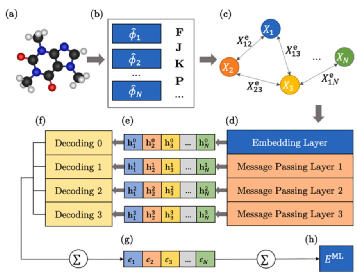
图6 OrbNet图解图
量子化学中基于轨道图的学习模型的最新发展促使我们在图神经网络学习体系中采用基于轨道图的固态材料系统。为了充分考虑晶体中的轨道相互作用,我们最近提出了GNN来有效地学习固体材料的哈密顿量。材料哈密顿量代表了任何物质中原子轨道之间的相互作用,它提供了控制固体化合物中结构-性质相关性的所有基本元素。在这项工作中,我们提出了几种不同的图形卷积网络,目的是预测无机材料的带隙。通过结合两种不同的轨道特征(每个轨道的信息和轨道间的相互作用),我们的模型对530个半-Heusler化合物进行了训练,与浅层学习模型相比,该模型对金属-非金属分类任务具有良好的预测精度。这项工作是继续开发用于固体材料深度学习的轨道图的基本步骤。
最近,一种轨道图卷积神经网络(OGCNN)被提出以部分考虑晶体材料的原子轨道信息。该结构如图7所示。该模型基于CGCNN模型,将原子轨道间的成键信息编码到轨道场矩阵(OFM)表示中,其中使用组成原子的电子组态和被邻近原子覆盖的固体角度作为附加信息。然后将节点和改进的边缘特征传递给CGCNN框架以进行单值预测。由于该框架中新的边缘特征向量比CGCNN中的特征向量大得多,因此前端改进的网络在形成能量和带隙方面都比CGCNN具有更强的学习能力和更好的预测性能。此外,由于在体系结构中使用了更多关于绑定的信息,因此增强了节点之间的通信。
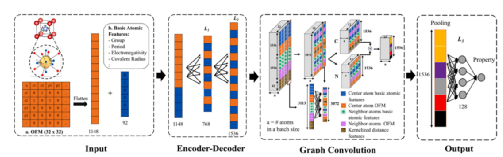
图7 OGCNN图解图
总结展望
尽管还处于起步阶段,但基于图形的深度学习框架已经通过识别结构与性质的相关性和进行有效的性质预测,展示了它们在设计和发现功能材料方面的创造性作用。通过用图形表示物质系统和适当地设计消息传递策略,深度学习体系结构在增强物质系统的学习的物理原理方面获得了新的灵活性、效率和可靠性。在达到基于图表的深度学习框架可以轻松应用于各种不同材料系统的阶段之前,社区需要解决几个关键挑战。首先,从CNN的结构来看,GCNN中的端到端学习策略通常需要大量的训练样本。在图像识别领域,如果没有ImageNet的引入,深度学习的广泛应用是不可能的,ImageNet是一个包含数百万手动标记图像的大型数据集。对于固态无机材料,不幸的是,我们没有如此大量的标记数据(至少目前是这样)。为了解决这一问题,需要社区共同努力,以高精度创建大量具有标准格式的标签材料数据。此外,应该将领域知识纳入消息传递策略的设计中,以控制数据相对有限的学习模型的过度匹配。
另一个挑战与培训成本或分子和固态系统模型的可扩展性有关。例如,人们注意到,当处理较大的分子时,MPNN的性能显著下降,这归因于成对相互作用和完全连接的信息传递通道。密集图的消息传递阶段的单个步骤需要O(n[2]d2)个浮点乘法,其中n是节点数,d是节点向量的维度。对于超越小分子和晶体的大型材料系统来说,这在计算上是昂贵的。为了克服这一可伸缩性问题,基于图的学习框架的开发可以有效地推广到更大的图。正如MPNN的工作所指出的,在传入的消息向量上添加注意机制是一个有趣的探索方向,并且沿着这条路线在分子系统上已经有了一些应用。
在固态晶体材料领域,社区面临的一个剩余挑战是对电子结构和相关物理性质的可靠预测。现有的图学习框架对电子结构相关材料数量(如带隙和带边)的预测仍然不能令人满意,需要进一步改进。以图为中心的深度学习框架的未来发展依赖于超越真实空间的新颖架构设计,并考虑到原子系统的多层物质信息,如轨道、局部和全局对称性、拓扑和互易空间信息。最后但并非最不重要的是,将基于图的最大似然学习应用于材料科学的一个重要方面是将基于小图形和规则图的学习模型扩展到大型和不规则材料系统的应用,如具有点和线缺陷的固体、表面和界面结构、分子-固体杂化系统等。那些在ML的其他领域正在积极发展的技术,如迁移学习和图形注意,值得更多地关注,以确定它们在这一新兴且令人兴奋的研究领域中的角色。
注:本公众号推送内容以交流学习为目的,并非商业用途,如有侵权,请联系协商处理。欢迎各位专家学者投稿,分享Ai+材料最新科研成果!
投稿邮箱:zyj1047587695@qq.com
群聊:材料与人工智能-学习交流群
加群方式:添加编辑微信:zyj1047587695,请备注:单位-姓名-研究方向,由编辑审核后邀请入群

材料与人工智能
斗转星移,光阴飞逝,AI使世界变化之快,如过隙白驹,让我们目不暇接。一种难以厘清的压迫感隐约而至。科学研究方式与生态是否处于大变革前夜?我们冥思,我们求索,为此创办此公众号,与大家分享和讨论AI在材料科学领域的最新进展、问题和挑战。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢