
编译 | 曾全晨
审稿 | 王建民
今天为大家介绍的是来自范德堡大学研究团队的一篇关于药物发现的论文。在计算辅助药物发现中,通过化学结构训练定量构效关系模型以预测生物活性。尽管将图神经网络应用于该任务取得了一定成功,但重要的化学信息,如分子手性,被忽略了。为了填补这一重要的空白,作者提出了分子核图神经网络(MolKGNN)用于分子表示学习,具有SE(3) /构象不变性、手性感知和可解释性的特点。

开发新药物是耗时且昂贵的,例如,开发一个抗肿瘤药物cabozantinib需要8.8年和19亿美元才能上市。为了协助这个过程,研究人员广泛使用计算辅助药物发现(CADD)。在CADD中,已经开发了几种数学和机器学习方法来建立定量构效关系(QSAR)模型,以根据分子的几何结构预测其生物活性。最近,图神经网络(GNN)已成功应用于许多领域,例如社交网络和推荐系统。由于分子本质上可以被视为以原子为节点、化学键为边的图,因此GNN自然而然地被采用来进行图分类,即根据分子的几何结构预测其生物活性。用于图分类的典型GNN架构始于通过传递邻域信息来提取节点表示的编码器,然后通过汇集操作将节点表示集成到图表示中,再将其输入分类器以预测图类别。
尽管应用于分子表示学习的GNN模型具有潜力,但现有的GNN模型要么盲目地遵循信息传递框架而不考虑图上的分子约束,要么无法整合手性,或者缺乏可解释性。为了填补这一关键空白,作者开发了一种名为MolKGNN的新型GNN模型,具有SE(3) /构象不变性、手性感知性,并提供一种可解释性的形式。
模型部分
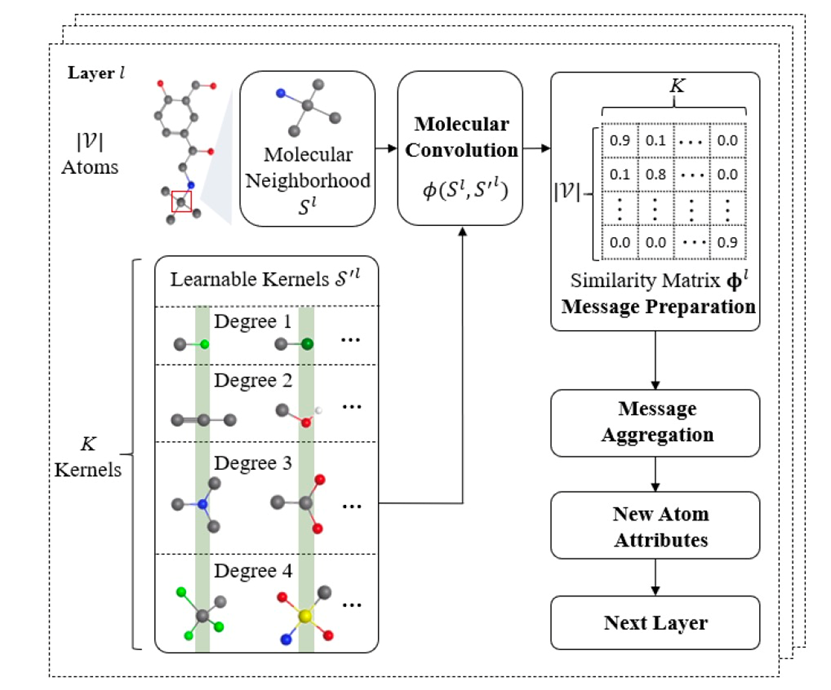
图 2
作者提出的MolKGNN的框架如图2所示,MolKGNN通过递归地执行分子卷积和消息聚合来学习每个分子的表示。在分子卷积中,作者设计可学习的分子卷积核,以捕捉每个节点/原子的化学上有意义的子图模式。具体而言,模型计算每个原子与其邻域之间的相似度得分,并将所得分数视为新的原子特征,这本质上描述了原子化学性质与内核编码模式之间的距离。然后,在消息聚合中,利用特征传播来聚合邻域的相似度得分,以进一步捕捉每个原子的化学环境。这两个模块交替进行,逐渐扩大感受野,以捕捉更高阶的化学模式。
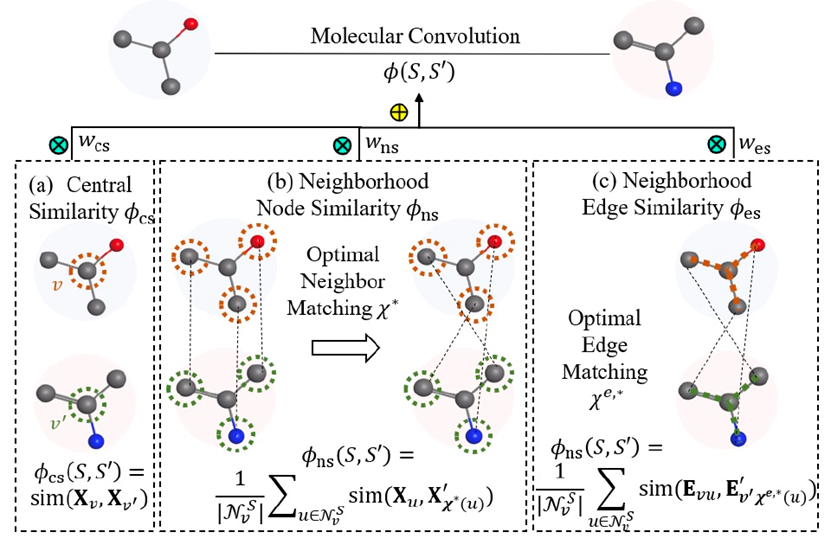
图 3
在二维图像中,卷积操作可以被视为计算图像补丁与图像卷积核之间的相似性。较大的输出值表示更高的视觉相似性模式,例如边缘、条纹、曲线。受此启发,作者设计了一种分子卷积,当分子邻域和卷积核在化学上更相似时输出较高的值。模型从中心相似性、邻域相似性和边缘相似性三个不同方面量化相似性,计算方式如图3所示。首先,模型通过计算原子与卷积核中的中心节点的相似性来捕捉原子本身在子图中的化学性质。除了中心节点外,原子的化学性质还受其邻域上下文的影响,模型进一步量化子图和卷积核之间的以下相似性:1)子图中的相邻节点和卷积核中的相邻节点之间的相似性,以及2)子图边和卷积核种边之间的相似性。由于数据集中原子化学键的个数普遍小于等于四个,且受手性影响(仅有一半的匹配方式合理),在实际计算过程中,子图中原子和卷积核中原子匹配方式最多只有12种(4!的一半),并且边匹配过程不需要单独计算,因此从计算量角度可行。
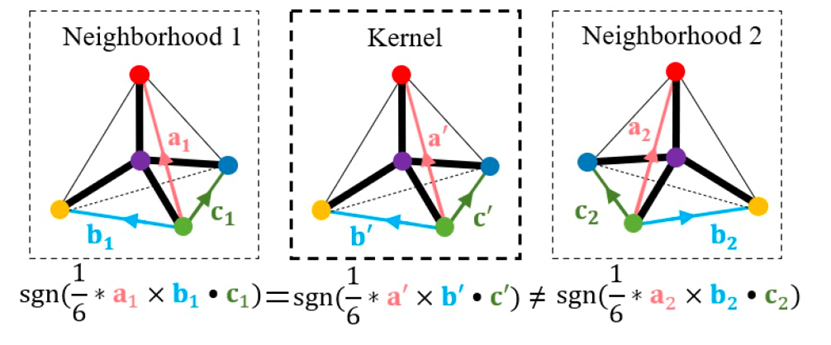
图 4
在通过量化上述三种不同方面的相似性来捕捉每个原子周围邻域子图的化学信息模式后,模型仍然是对手性无感知的。然而,手性是分子生物活性的关键决定因素,这促使模型需要在下一步对分子的手性进行表征。模型对此操作具体地说,给定一个原子S的邻域子图,形成如图4所示的四面体,其中四个唯一的相邻原子是NSv = {u1, u2, u3, u4},选择u1作为锚点邻居(以图4中绿色点为例),来定义四面体的三个共边边界。随后利用向量叉积公式确定方向,这个过程类似SMILES中如何确定手性的方法。倘若子图和卷积核方向相同,不改变图3中分子卷积的结果,倘若方向相反,则取负。模型由此来获得手性感知能力。
上述分子卷积仅能捕捉原子周围1跳邻域中嵌入的化学模式。为了进一步发现多跳邻域中嵌入的化学模式,作者利用消息传递并直接聚合计算得到的邻域相似性。在经过分子卷积和消息传递的L层之后,最终的原子表示描述了每个原子距离为L跳的化学模式。实际模型可以通过递归交替执行分子卷积和消息传递操作,获得化学模式的表示。
实验部分
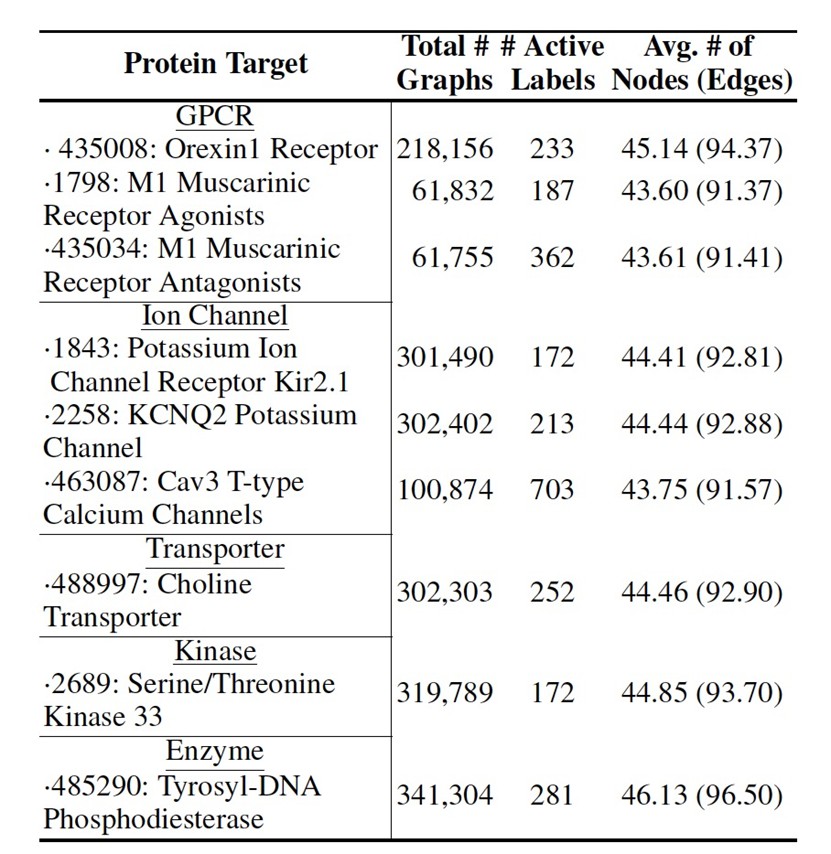
表 1
实验数据来自PubChem,它是由美国国立卫生研究院(NIH)支持的数据库,包含数百万种类似药物的分子的生物活性数据,通常来自高通量筛选实验。然而,来自PubChem的原始初筛数据具有很高的假阳性率。作者使用来自PubChem的九个高质量的高通量筛选实验对我们的模型进行基准测试,这些实验涵盖了药物发现中所有重要的蛋白质靶点类别,摘要见表1。这些数据集具有大规模数据、高度不平衡的标签和多样的蛋白质靶点特征。
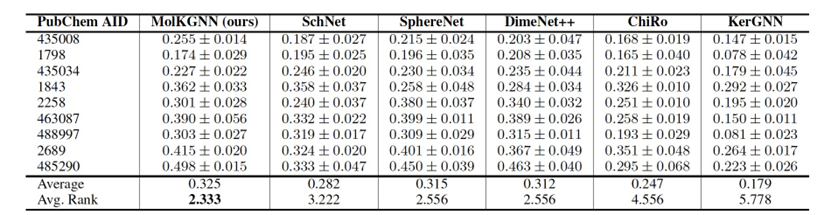
表 2
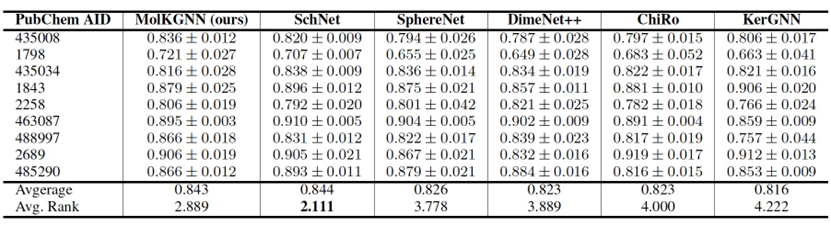
表 3
从表2中可以看出,MolKGNN在高决策阈值下恢复活性分子方面取得了优越的结果。这证明了MolKGNN在实际场景中的适用性。此外,可以发现MolKGNN在AUC方面的表现与其他GNN相当(见表3),这表明它在除药物发现以外的一般情况下也具有潜在的适用性。值得注意的是,在这两个表中观察到不同模型的排名不同。这表明一个AUC表现良好的模型在特定的假阳性率区域可能表现不佳。此外,这也凸显了所提出的模型在应用相关度量方面表现良好,并显示了其实际意义。
结论
在此项研究中,作者引入了一种新的图神经网络模型MolKGNN,用于解决QSAR建模问题。MolKGNN采用了一种新设计的分子卷积,其中将分子邻域与内核进行比较,并输出相似性分数。作者使用经过数据集进行评估,这些数据集包含来自不同蛋白质靶点类的实验高通量筛选(HTS)数据,评估结果展示了MolKGNN在药物发现中的实际价值,同时也提供了用于比较的一般度量(AUC)的性能指标。
参考资料
Liu, Y. L., Wang, Y., Vu, O., Moretti, R., Bodenheimer, B., Meiler, J., & Derr, T. (2023, June). Interpretable chirality-aware graph neural network for quantitative structure activity relationship modeling in drug discovery. In Proceedings of the AAAI Conference on Artificial Intelligence (Vol. 37, No. 12, pp. 14356-14364).
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢