
编译 | 曾全晨
审稿 | 王建民
今天为大家介绍的是来自Nilah Ioannidis的一篇讨论深度学习模型在基因序列应用的论文。基因组深度学习模型可以直接从DNA序列预测全基因组的表观遗传特征和基因表达水平。尽管当前的模型在从参考基因组预测不同细胞类型的基因表达水平方面表现良好,但它们在解释个体间由于顺式调控基因变异而导致的表达变异能力仍然未被充分探索。在这里,作者对四种最先进的模型进行了个体基因组与转录组数据配对的评估,发现在解释个体间表达变异方面的性能有限。

随着深度学习的快速发展和用于训练的数据集的增长,最近在直接从参考基因组序列中预测基因表达水平、3D基因组折叠以及表观遗传特征,如转录因子结合、组蛋白修饰和染色质可及性等取得了成功。这些基因组深度学习模型是通过来自各种细胞类型和细胞背景的全基因组数据进行训练的,并已被证明能够学习到输入DNA序列中具有生物学相关的调控模体。目前的序列到表达模型可以根据每个基因起始位点周围的参考基因组序列来解释基因组中不同基因的表达差异。然而,将这些模型应用于个人基因组的序列,以解释不同个体之间的基因表达差异(图1a),在很大程度上尚未被探索。在这里,作者评估了四种最先进的模型 Enformer,Basenji2,ExPecto和Xpresso,使用Geuvadis的成对全基因组测序和RNA测序数据(n = 421),并显示出当解释个体间基因表达变异时,模型性能受到限制。当模型确实捕捉到调控变异时,仅对有限的一组基因来说,它们常常无法准确捕捉这种变异对表达的正确影响方向。
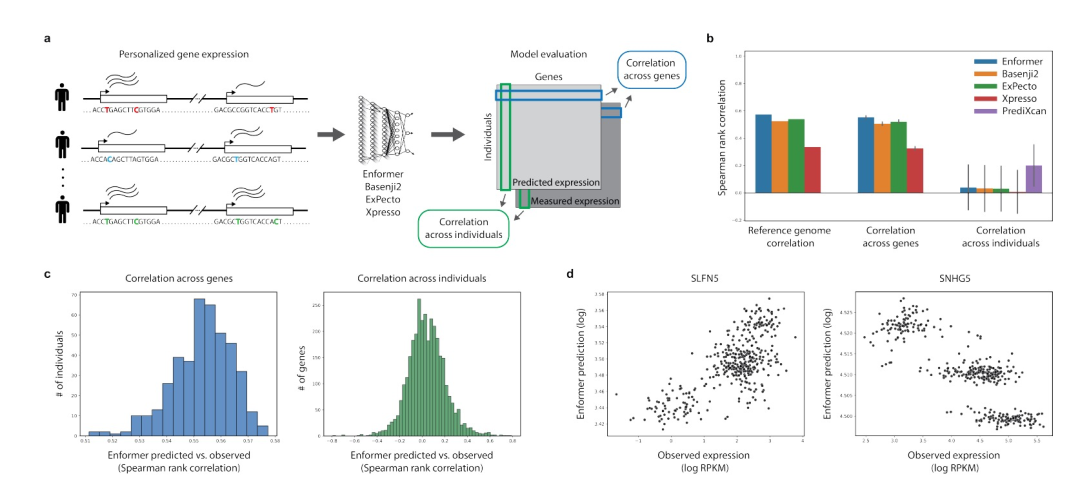
图 1
为了在个人基因组变异上测试这些现有的序列到表达模型,作者使用了来自Geuvadis的RNA测序数据,这些数据是在淋巴细胞母细胞系(LCLs)上测量的,并且与1000个基因组计划中的421名个体的全基因组测序(WGS)数据相配对。作者关注3259个基因,这些基因在Geuvadis的表达定量位点(eQTLs)分析中至少被识别出一个统计显著的遗传关联,其中一个顺式变异体的基因型可以预测个体之间的基因表达变异。作者通过将每个个体的单核苷酸变异(SNVs)插入到每个基因转录起始位点(TSS)周围的参考序列中,构建了每个个体的个人输入序列。然后,作者使用所有四个模型计算每个个体以及参考基因组序列的基因表达预测结果。对于每个模型,我们使用输出表达预测轨迹,其与用于Geuvadis测量的LCLs最相似的细胞类型相对应。为了确保所选择的模型输出确实与LCLs中基因表达的预测相关,对于每个基因,作者将使用参考序列的模型预测与其在Geuvadis数据集中的中位数表达水平进行比较(图1b)。可以发现Enformer的预测与观察到的表达水平之间的Spearman秩相关系数为0.57,Basenji2为0.52,ExPecto为0.54,Xpresso为0.33,这表明这些模型解释了LCLs中基因表达变异的显著部分,与先前的研究结果相似。
对于每个模型,作者使用个性化序列作为输入,计算了两个额外的指标。首先,对于每个个体,计算一个交叉基因相关性,将使用该个体的个人输入序列预测的前述3,259个基因的表达水平与该个体中这些基因的观察表达水平进行比较。类似地,对于每个基因,作者计算一个交叉个体相关性,将在所有421名个体中使用预测表达水平与其观察表达水平进行比较(请参见图1a)。可以发现,每个个体的交叉基因相关性与相应模型的参考基因组性能相似(图1b、c),Enformer的平均Spearman相关系数为0.55,Basenji2为0.51,ExPecto为0.52,Xpresso为0.32。然而,当对每个基因计算跨个体的相关性时,发现所有模型的跨个体相关性分布接近于零(图1b、c),这表明所有模型在解释个体间表达差异方面都存在困难。这个结果表明,目前最先进的序列到表达模型没有正确预测许多单核苷酸变异对基因表达的影响。
相比之下,针对每个基因分别使用附近变异剂量作为预测因子进行训练的正则化线性回归模型,即使限制在与Enformer相同的输入上下文(197kb)内,也能解释更多的个体间变异(图1b)。由于这种PrediXcan风格的模型并不试图学习可适用于训练集之外的新序列、变异体和基因的可推广序列特征,作者将这些模型包含在内,不作为竞争方法,而是作为数据集中每个基因可能学习到的基因表达的最低基线。这些PrediXcan风格模型的更高性能表明,常见的顺式调控变异体的效应无法被当前的深度学习模型所捕捉。
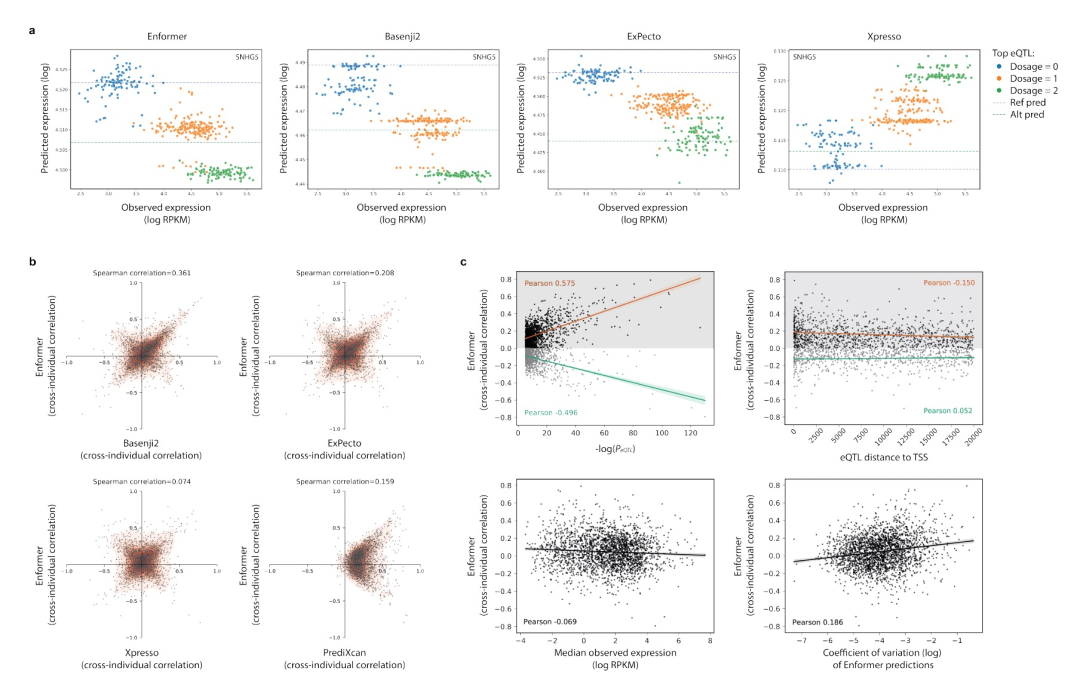
图 2
作者还发现,尽管所有模型的平均交叉个体相关性接近于零,但对于每个模型,都存在强正相关和强负相关基因的尾部分布(图1c)。例如,图1d显示了Enformer模型中具有强正相关和强负相关的样本基因。当在所有四个模型之间比较这些基因的预测时,可以发现模型在相关性的方向上常常互相产生分歧(图2a、b)。这个结果表明,对于任何给定的模型,对于负相关基因的遗传效应的方向的错误预测并不是由于模型在建模这些特定基因或其对应的变异体方面存在困难,而是由于这些类型的模型对变异体的效应进行归因时存在噪声。重要的是,作者发现,在给定基因的预测与观察到的表达之间,这四个测试模型在相关性的大小上更加一致,而不是在相关性的方向上(图2b),这表明它们在识别引起调控变异体方面比在这些变异体对表达的效应方向上更一致。
接下来,作者探索了基因表达遗传效应的预测方向是否对特定类型的基因更准确。首先,作者测试了在Geuvadis eQTL分析中具有强遗传关联的基因是否更有可能有正确预测的遗传效应方向,通过将每个基因的交叉个体相关性与距离TSS(转录起始位点) 20kb内最显著的eQTL的p值(图2c)、效应大小和最小等位基因频率进行比较。可以发现,具有强eQTL的基因在所有模型中往往具有较大的交叉个体相关性;然而,这些基因并不比负相关的交叉个体相关性更可能为正相关,这表明模型经常预测具有强遗传效应的基因的效应方向是错误的。
结论
总结起来,作者对四个最先进的序列到表达深度学习模型(Enformer、Basenji2、ExPecto和Xpresso)在个性化基因表达预测上的表现进行了分析,发现这些模型在根据个体间输入DNA序列的差异来预测给定基因在个体间的表达差异时表现普遍不佳。作者还发现,存在一些基因的预测表达水平与观察到的表达水平之间存在强烈的负相关性,这些基因的模型可能已经识别出了引起调控的变异体,但是错误地预测了其效应方向。先前关于变异体效应预测的评估主要集中在个体变异体效应的研究,例如eQTL研究或大规模并行记者基因测定实验(MPRA)。然而,MPRA缺乏内源基因表达的复杂基因组和染色质环境,而即使采用当前的精细定位方法,也很难在eQTL研究中确定原因变异体,导致与原因变异体在连锁不平衡中的变异体效应大小估计没有生物学意义。通过使用个人基因组序列来评估模型性能,作者的输入序列包括每个个体TSS周围的所有变异体,从而避免了因果变异体识别的问题。作者的关于方向性的结论与先前关于eQTL的测试结果一致,这些结果显示在预测个体变异体对表达的效应方向时表现较差,特别是对于远程eQTL。
参考资料
Huang, C., Shuai, R., Baokar, P., Chung, R., Rastogi, R., Kathail, P., & Ioannidis, N. M. (2023). Personal transcriptome variation is poorly explained by current genomic deep learning models. bioRxiv, 2023-06.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢