
编译 | 曾全晨
审稿 | 王建民
今天为大家介绍的是来自Jian Zhou的一篇的论文。为了了解基因组序列对多尺度三维基因组结构的影响,本文介绍了一种基于序列的深度学习方法,名为Orca,可以直接从序列中预测从千碱基到整个染色体尺度的三维基因组结构。Orca捕捉了包括染色质区块和拓扑联合域在内的结构的序列依赖性,以及与细胞类型特异性相关的各种类型的相互作用,包括CTCF介导的相互作用、增强子-启动子相互作用和Polycomb介导的相互作用。

了解基因组序列如何指导基因组在各种空间尺度上折叠成三维结构,对于解释基因组序列和基因组变异在正常和疾病状态下参与各种细胞过程将具有指导意义。这样的序列依赖性可能是多重的,因为有多个方面的三维基因组组织似乎对应于不同的机制。最显著的是,染色质区块通常在兆碱基尺度上观察到,具有特征性的交错式相互作用模式,其中区块A和区块B主要对应于表达活跃和不活跃的染色质,它们优先与相同区块进行相互作用。拓扑联合域(TADs)通常在100 kb到1 Mb的尺度上发现,具有常见的嵌套结构。尽管已知与基因表达活性和特定的组蛋白标记相关,但染色质区块大尺度组织的序列基础仍未解决。
在亚兆碱基尺度上,TADs的形成众所周知地依赖于CTCF序列基序,可能通过CTCF-凝聚素依赖的环状挤出机制实现。然而,多种类型的CTCF独立相互作用的序列决定因素,包括增强子-启动子相互作用和Polycomb诱导的接触,目前了解得较少,更不用说从序列预测这些相互作用了。
基于高通量染色质构象捕获(3C)的方法,全面记录了从千碱基到整个染色体尺度的各种基因组相互作用。这为开发机器学习方法识别基因组相互作用的复杂序列依赖关系提供了基础。学习跨空间尺度的3D基因组结构的序列依赖关系将重要地提供预测新序列影响的能力。从序列中预测多尺度3D基因组结构不仅可以预测任何序列变异的影响,还有助于理解3D基因组组织的新序列机制。深度学习序列模型已应用于基于基因组序列的各种生化和调控特性的建模。最近的研究已经在基于序列的子兆碱基3D基因组结构建模方面取得了突破,允许从基因组序列预测基因组相互作用,距离可达1 Mb。然而,尚未开发出预测涉及1 Mb以上序列上下文的大规模基因组组织的序列模型。这限制了预测大结构的能力,包括依赖更大序列上下文的染色质区块和局部结构。此外,缺乏大规模序列模型也限制了对大结构变异(SVs)的建模能力,这是最具影响力的基因组变异之一。
为了实现对由Hi-C类型方法测量的基因组体系结构的所有尺度进行建模,作者开发了Orca,这是一个多尺度序列建模框架,可以从序列中预测基于3C数据的千碱基到整个染色体尺度的3D基因组结构。Orca可以预测多种结构,包括TADs(拓扑关联结构域),染色质A/B区块,Polycomb介导的相互作用以及启动子-增强子相互作用。此外,该方法可以预测基因组中任意一对序列之间的染色体内和染色体间相互作用。
模型结构
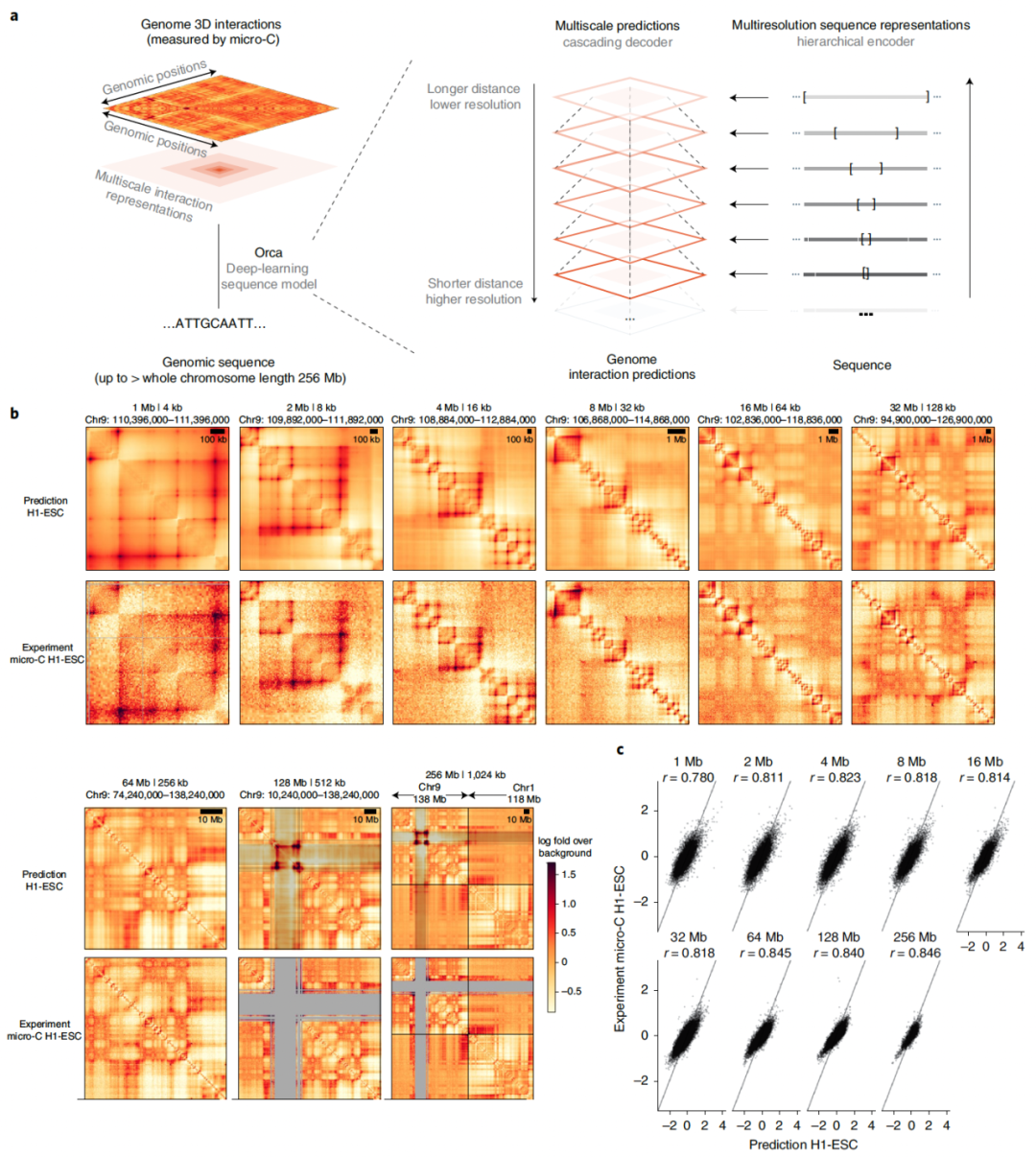
图 1
基于序列的多尺度3D基因组相互作用预测。多尺度的染色质组织展现出不同的特征,并可能涉及多种机制,使用深度学习从单个核苷酸到整个染色体的所有尺度捕获序列依赖关系是一项前所未有的挑战。为了解决这个挑战,作者首先开发了一种多尺度深度学习序列建模框架,名为Orca。为了在整个基因组距离尺度范围内进行预测,设计了一种类似“缩放”的级联预测机制,使得能够从超长距离相互作用预测到更短距离的相互作用,共有九种不同的分辨率(例如,在1-Mb距离上为4 kb,2-Mb距离上为8 kb,128-Mb距离上为512 kb)。由于Hi-C类型数据通常通过多分辨率矩阵表示,而更长距离的大尺度结构通常基于更稀疏的测序读数检测,因此只能以较低的分辨率测量,因此设计了在不同分辨率下建模多尺度结构,以适应这些数据类型。
模型架构由一个分层多分辨率序列编码器和一个级联多级解码器组成。编码器将最多256-Mb的序列作为输入,并生成从4 kb到1,024 kb的九种不同分辨率的越来越粗粒度的序列表示。多级解码器在顶层预测最多256-Mb距离的相互作用,这比最长的人类染色体chr1还要长,并在底层预测1-Mb距离内的4-kb分辨率相互作用。通过使用多染色体输入,还允许在32–256-Mb级别进行染色体间相互作用。为了实现对大尺度染色体序列的深度学习模型训练和推断的扩展,提出了一种用于增加内存效率的水平检查点技术,允许在内部表示大小远远超过GPU内存限制时仍能训练模型。
Orca序列模型是在H1胚胎干细胞(H1-ESCs)和人类包皮纤维母细胞(HFFs)的数据集上进行训练的,这些数据集是迄今为止分辨率最高的数据集之一。编码器和解码器在三个阶段联合训练,其中在较早阶段训练的编码器被冻结并在后续阶段训练中使用。最终的模型在九个不同尺度上(图1a-c)预测从1到256 Mb。每个模型包括1-Mb,1-32-Mb和32-256-Mb模块,可以一起或分开使用,以提供灵活性:1-32-Mb模型是主要的模型,在大多数应用中具有高精度和灵活性;32-256-Mb模型对于预测染色体尺度和染色体间相互作用最有用;1-Mb模型对于快速筛选大量变异体的局部基因组相互作用效应很有用。预测的相互作用矩阵得分表示相对于基于距离的背景得分的对数倍数,其中背景得分(通常称为预期得分)是相同基因组距离下的平均归一化接触得分。在测试染色体上,该模型在H1-ESCs上在所有尺度上与实验观测保持0.78-0.85的Pearson相关性,并且在HFFs上保持0.73-0.79的Pearson相关性。染色体间相互作用的预测与0.47-0.74的相关性(64-256-Mb水平)。
从序列预测的多尺度结构变异(SV)对3D基因组的影响
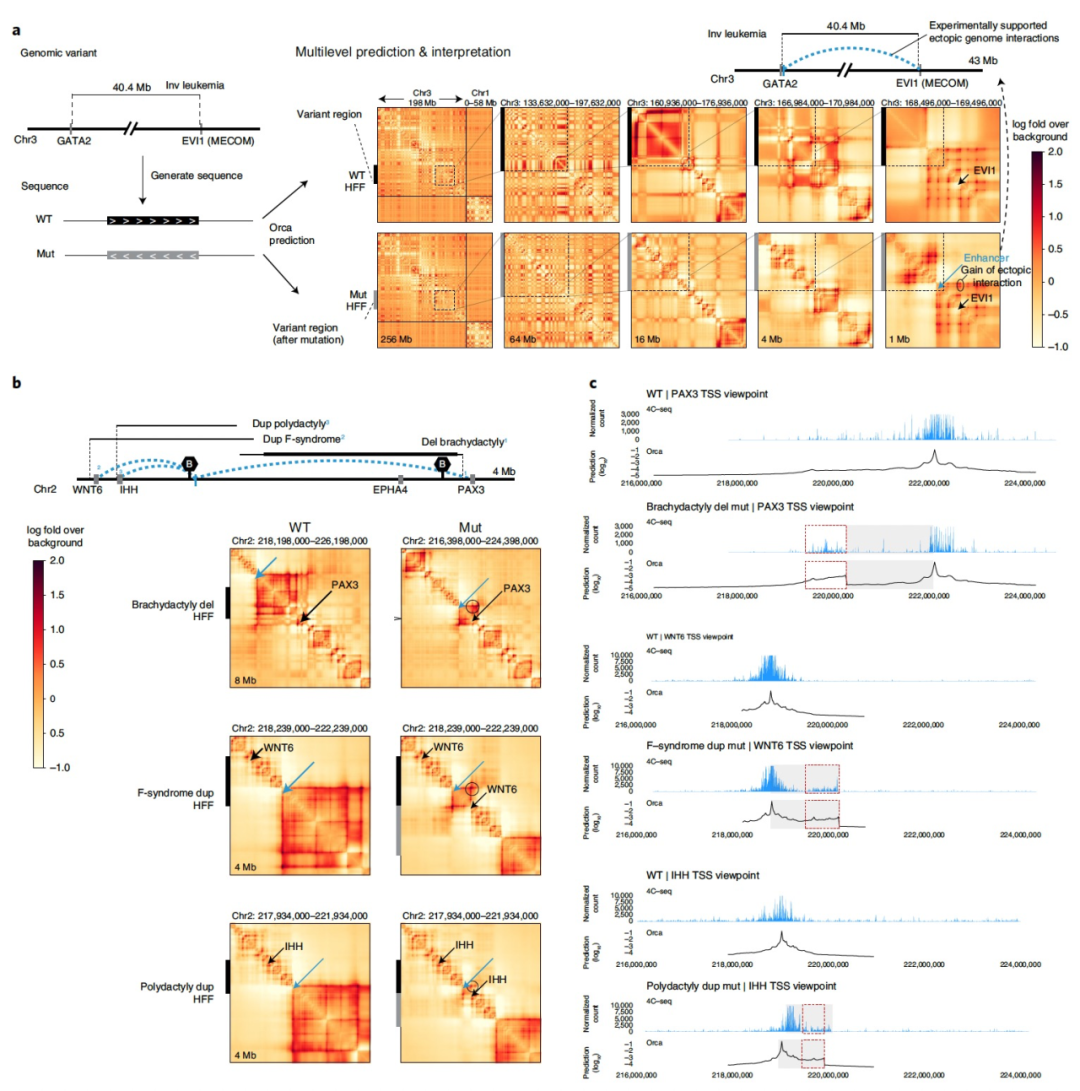
图 2
由于Orca模型可以准确地预测新的未见序列上的基因组相互作用,因此在预测基因组变异效应时它们可能特别有用。特别是,因为Orca允许非常大的序列输入(256 Mb,大于最长的人类染色体chr1:249 Mb),它可以预测几乎任何大小的变异效应。要预测任何变异的基因组结构影响,可以计算重建携带变异的染色体序列,并将预测与参考序列的预测进行比较。也可以以类似的方式预测同一单倍型上多个变异的联合效应。
首先测试了转座子介导的2-kb TAD边界元素插入到各种基因组位置的SV效应预测,这些插入已经通过原位Hi-C测量。计算了每个插入位点的隔离得分变化,并与预测的变化进行比较。在14个插入位点中,对于H1-ESC模型,Orca在隔离得分变化方面获得了0.89的余弦相似度分数,对于HFF模型获得了0.76的分数(P < 1 × 10−4)。此外,Orca的预测重新展现了所有三类插入效应,包括新边界的形成、现有边界的加强以及没有区域级别的影响。因此,实验中的Hi-C测量结果与Orca对这些插入的基因组结构影响的预测高度一致。为了评估模型在预测多种SVs影响方面的能力,对各种大小从0.3 kb到80 Mb的SVs的影响进行了预测,c并测得了其基因组结构影响(Fig. 2)。首先展示了多尺度结构影响预测,使用一个大的40.5-Mb倒位突变作为示例,该突变被认为是急性髓系白血病的潜在原因,并在从整个染色体视图到EVI1邻近断裂点的五个不同层次上显示了预测结果(Fig. 2a)。预测展示了染色体组织的大规模重组以及与断裂点相邻的染色质区块和TADs的影响,包括最细粒度层次上EVI1启动子与GATA2增强子之间的相互作用增加,这已经经过实验确认。接下来,Orca的预测应用于分析一个复杂的区域,该区域存在多个删除、倒位和复制变异,其大小从0.9 Mb到1.8 Mb,导致了几种不同的肢体畸形表型:短指畸形、F综合征和多指畸形。Orca预测显示,通过不同的结构变化,所有这些疾病SVs都会在同一增强子区域上引起三种不同基因(PAX3、WNT6和IHH)之间的新联系(Fig. 2b、c)。这些预测也与基于环状染色体构象捕获(4C)实验的先前实验数据完全一致。
基序(motifs)是细胞类型特异性局部基因组相互作用的基础
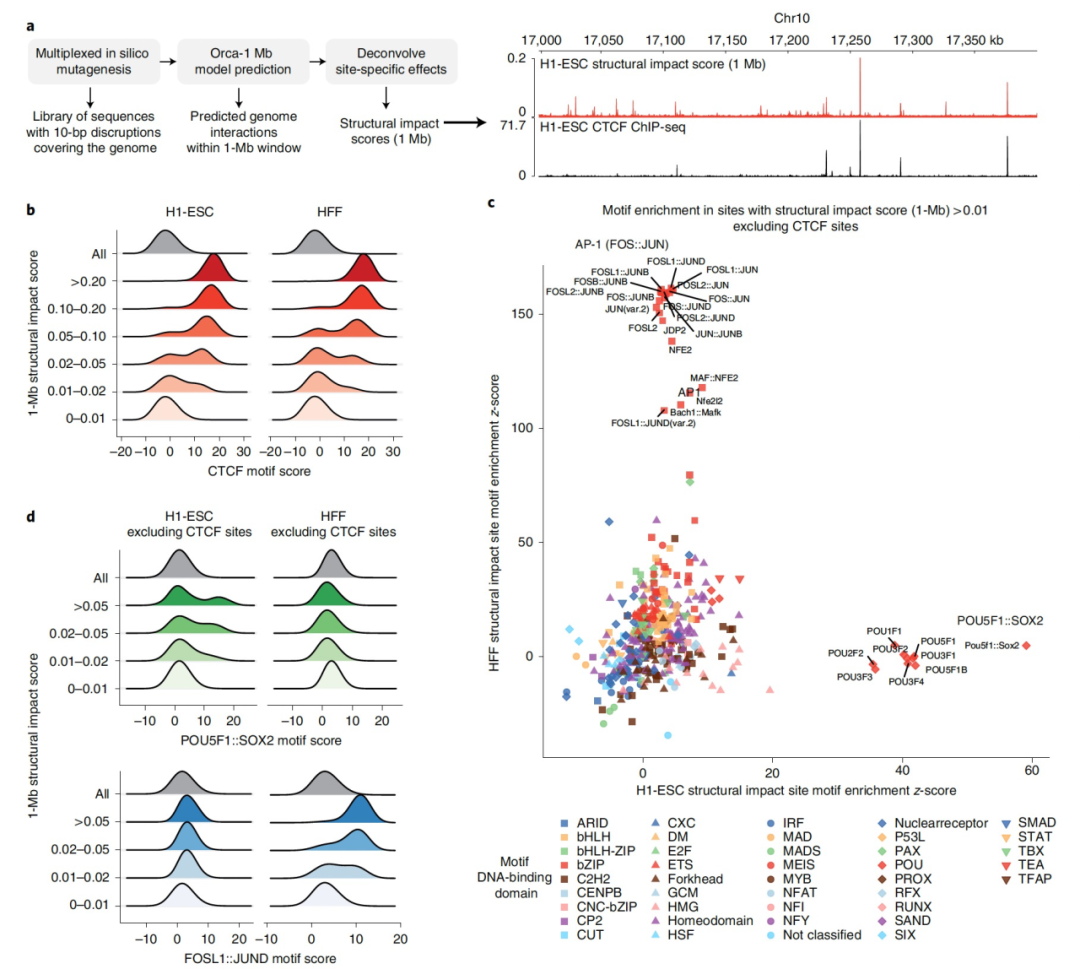
图 3
模型能够直接从基因组序列预测多尺度的3D基因组结构,使其能够作为一个“基因组观察台”来探索深度学习模型学习到的3D基因组组织的序列决定因素。这种计算方法具有在大量序列上进行“虚拟遗传筛选”的能力,并且在序列设计方面几乎具有无限的灵活性。在这里,作者设计了多个筛选策略,用于解析局部(1-Mb)和区段级(32-Mb)组织的序列基础,从而揭示了不同的序列依赖性。为了发现亚兆碱基尺度基因组结构的序列依赖性,如TAD、亚TAD和启动子-增强子相互作用,作者设计了一种多重的体外突变方法,用于筛选在1-Mb距离内导致“局部”结构重构的序列破坏(图3a)。这种多重方法将多个位点破坏突变引入相同的1-Mb序列中,以加速近碱基级别的筛选,。此外,每个10碱基位点在三个不同的序列中被破坏,每个序列都有一个随机的破坏位点集。通过利用突变的稀疏性,使用最小的1-Mb结构影响分数(破坏位置与1-Mb窗口内的所有其他位置之间的平均绝对对数折叠交互变化)对具有相同破坏位点的三个序列进行了解析。每个序列都有独立的随机破坏,从而可以过滤掉仅由特定突变序列引起的低概率事件。
采用这种方法,对染色体上所有的10碱基对序列进行了筛选,这些序列的破坏会对结构产生影响。与CTCF在TAD级结构组织中的核心作用一致,对于H1-ESCs和HFFs,大多数10碱基对位点(>88.9%)在1-Mb结构影响分数最强的层次(>0.1,<0.015%的基因组)与CTCF基序重叠(图3b),>95.1%的位点与CTCF基序的距离在200-bp以内,而仅有<1%的位点在200-bp距离内缺乏CTCF基序,而与全基因组背景(64%)相比,CTCF基序是不缺乏的。这表明,最强影响位点主要由CTCF解释。然而,并非所有CTCF基序都被预测为具有强大的结构影响(只有约1%的具有CTCF基序对数概率>10的位点具有结构影响分数>0.1);因此,CTCF基序并不是唯一的决定因素,该模型利用更复杂的序列依赖性来进行准确预测。尽管1-Mb结构影响分数最强的位点主要与CTCF相关,但在中等影响分数范围(0.01-0.1,约0.2%的基因组)中,非CTCF转录因子基序也显著富集(排除了与任何附近CTCF基序或结合位点相邻的位点)(图3c,d)。此外,与CTCF基序依赖性相比,这些预测会影响基因组结构的非CTCF基序在细胞类型间具有非常强的特异性(图3c,d)。H1-ESC被预测为最容易对POU5F1::SOX2二聚体基序和POU家族基序的破坏做出响应,而HFF对AP-1(FOS::JUN)基序的破坏非常敏感。这种细胞类型选择性与POU5F1和SOX2在胚胎干细胞中的基因调控作用以及AP-1在成纤维细胞中的作用一致。单个POU5F1::SOX2或AP-1基序的破坏可以导致H1-ESC和HFF细胞模型中预测的基因组相互作用的消失。这些结果表明,细胞类型特异性转录因子介导局部相互作用,并可能通过空间组织影响转录。
结论
Orca是一个基于基因组序列的序列模型框架,用于全局预测从千碱基到整个染色体的空间尺度下的3D基因组组织。它允许预测任何基因组变异,包括大规模结构变异和拷贝数变异的基因组结构影响。Orca准确地重新演示了之前进行实验研究的变异的结构影响。通过仅需要序列即可快速分析大量变异的潜力,它有助于加速对健康和疾病中结构变异作用的研究。除了实现规模上的预测变异效应外,这些捕获3D基因组相互作用结构序列依赖性的序列模型为通过虚拟基因屏幕探索基因组相互作用的序列水平机制提供了工具。
参考资料
Zhou, J. Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale. Nat Genet 54, 725–734 (2022).
https://doi.org/10.1038/s41588-022-01065-4
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢