

作者提出了一种灵活的、对数据无限制的强化学习框架,用于基于结构的从头药物设计(A flexible data-free framework for structure-based de novo drug design with reinforcement learning)。这是一种新的基于蒙特卡罗树搜索和3D分子结构的框架,因此称为3D-MCTS(Monte Carlo tree search)。与流行的以原子为中心的方法不同,3D-MCTS采用基于片段的分子编辑(molecular editing)策略。从小分子药物中采集的片段在预定义的反合成规则下重组,提高了药物的相似性和可合成性,克服了基于原子的方法的固有局限性。利用多线程并行仿真和基于实时能量约束的修剪策略(pruning strategy),3D-MCTS实现了显著的效率。实验结果显示了3D-MCTS在探索化学空间方面的优越能力,其框架的灵活性使得在此过程中很容易纳入领域知识,从而能够生成具有理想药效团和增强结合亲和力的分子,突出了其在各种药物设计场景中的潜力。
背景
基于结构的分子生成方法已经证明了它们在模拟配体和受体之间的几何和能量互补方面的潜力,从而促进了具有良好结合亲和力和靶特异性的分子的设计。尽管引入了分子生成的深层生成模型,但部分与化学直觉相矛盾的原子生成范式限制了生成分子的有效性和合成可及性。此外,深度学习模型对大规模结构数据的依赖性阻碍了其在不同目标间的适应性。
方法
作者基于MCTS算法开发了一个通用的基于片段的分子生成框架,以生成在几何形状和能量方面与特定蛋白质口袋匹配的3D分子。由于生成的分子依赖于修饰分子的先前状态,将片段生长过程数学建模为马尔可夫决策过程(MDP)。MDP的形式表示为M = (S, A, F, R),其中S表示状态空间。对于三维分子生成任务,分子的状态由其拓扑结构和三维构象决定,可以表示为S = Stop × Sconf_l × Sconf_g。其中,Stop表示分子的拓扑空间,Sconf_l和Sconf_g分别表示局部构象空间(由分子内的键长、键角和二面角决定)和全局构象空间(由分子与蛋白质口袋的相对空间位置决定)。
A表示动作空间(即修饰分子的方式)。与之前使用SMILES字符串进行二维分子生成的研究不同,在动作选择过程中操纵了蛋白质口袋中分子的拓扑结构和构象。由于3D-MCTS是基于片段智能生成的,因此根据片段来操纵分子。具体来说,3D-MCTS执行了三种不同的动作将分子状态从st转变为st+1。
首先,确定取代位置,其中所有与氢原子相连的重原子都被视为潜在的取代位置,以简化处理。其次,从片段库中选择合适的片段,并将其附加到上一步选择的重原子上。在构建片段库时,考虑了药物相似性和可合成性,使用BRICS规则将DrugBank数据库中的小分子药物片段化,并计算每个片段的频率(图1a)。然后,考虑到它们的药物相似性和合成可及性,选择了前50个最频繁出现的片段作为片段库。BRICS方法识别和切割分子中逆向合成的相关键,根据切割部位的化学环境来标记原子。这种方法定义了一套连接原子的规则,确保了合成分子的可行性。根据重组规则选择符合条件的片段,并将它们连接到上一步选择的原子上。最后,以15度的间隔旋转了新引入的可旋转键,产生了多个键构象,并保留了能量较低的构象。
F:S × A→S为状态转移函数。在片段链接过程中,状态转移是确定性的,因此,对于任何给定的状态-动作对,p(st+1|st,at) = 1。也就是说,通过进行修饰操作,当前分子一定会跃迁到具有修饰结构的下一个状态。
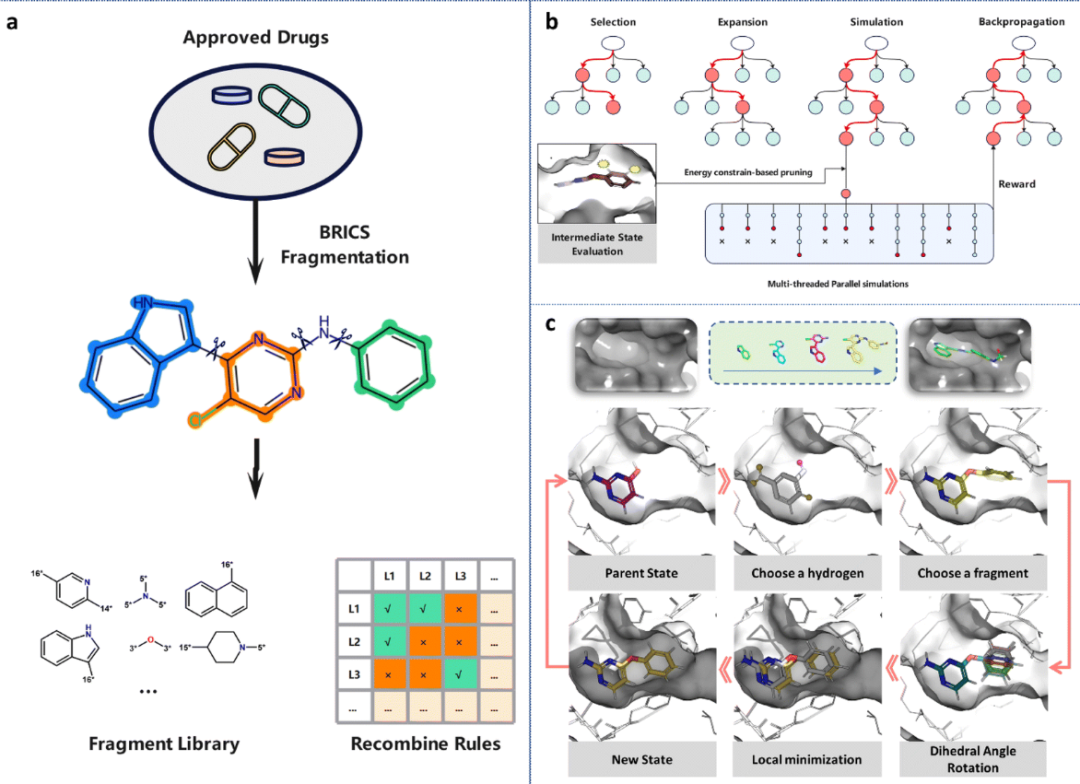
图1 3D-MCTS结构图
在一般的基于树的搜索中,应该评估每个节点及其后代,直到找到最终解决方案。然而,对于节点数量呈指数增长的任务,这种暴力搜索方法被证明是低效的。然而,通过MCTS(蒙特卡罗树搜索),一个机器学习算法通过专注于有利的后代节点而较少关注其他节点,可以探索最少数量的节点来获得最优解。MCTS采用树形结构来模拟和评估每一步中每个动作的值,同时利用先前估计的动作值来指导搜索过程,以获得更高的奖励。
首先,从根节点开始,从所有子节点中迭代选择最佳节点,直到到达叶节点(未探索的节点),使搜索过程能够快速收敛,同时避免陷入局部最优状态。为了降低陷入局部最优的风险,采用随机选择策略。扩展根据上面提到的动作空间A扩展所选择的叶节点。从一个未探索的叶节点开始,迭代生成下一个分子状态,直到满足终止条件。随机策略是MCTS中最常用的策略。与整个化学空间相比,在几何形状和能量方面与特定蛋白质口袋匹配的分子分布在一个非常有限的区域。
然而,使用完全随机的模拟策略会在无效空间中消耗大量的搜索时间,特别是对于需要探索拓扑和构象空间的3D分子生成任务。为了解决这个问题,3D-MCTS采用了多线程并行模拟方法,其中,对于每个选择的状态,在迭代过程中同时生成多个状态(图1b)。在模拟过程中,一种基于实时能量约束的修剪策略开始发挥作用,以评估分子中间状态与目标的匹配程度,丢弃能量不利的模拟路径(图1b)。利用Vina分数消除能量上不合理的分子构象,从合理状态中随机选择下一个状态。该方法有效地将搜索空间限制在具有合理能量的区域,并利用MCTS的随机性,避免了遇到局部最优的风险。当满足终止条件时,计算奖励值并随后沿着路径传播,以更新访问节点的状态。在标准的3D-MCTS中,奖励对应于在模拟路径中遇到的最佳Vina分数,并设定以下终止标准:
(1)原子碰撞:当体系中任何非键原子之间的距离小于其共价半径之和时,即认为发生了碰撞。
(2)不利能量:如果新片段的引入导致Vina分数增加,则认为该片段对分子与口袋的整体配合有不利影响。为了避免局部最优,提供了2 kcal mol−1的能量缓冲,使得只有当Score(st+1)−2 kcal mol−1 > Score(st)满足时,st+1才符合终止标准。
(3)类药物性质:观察到大多数分子的类药物性质随着分子体积的增大而恶化。因此,使用类药性作为终止准则可以防止搜索算法盲目地增加分子体积以获得更好的Vina分数。
(4)分子量:当分子量超过500时,st+1符合终止标准。
作者还引入了基于药效团的领域知识。与标准搜索相比,3D-MCTS算法中引入的药效团约束主要影响两个过程:(1)每个分子的奖励定义为Vina分数和特征计数的归一化平均值。在选择阶段,满足药效团模型的节点更有可能被选择。(2)在仿真过程中,特征计数作为选择的输入,倾向于满足药效团模型的状态。
结果
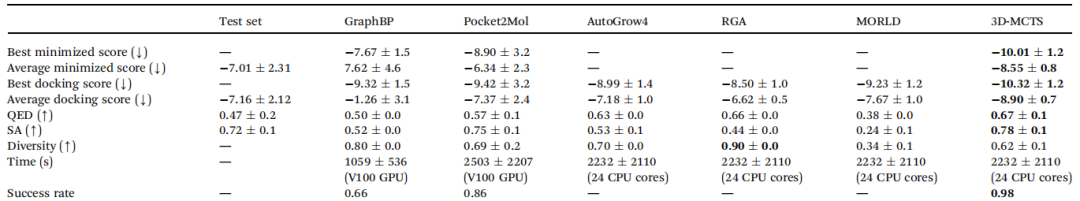
作者将3D-MCTS与一些具有代表性的方法进行了比较。表1列出了:
(1)最优最小分数、平均最小分数、最优对接分数、平均对接分数(以上四个分数越低越好);
(2)类药性(QED)SA分数(越高说明分子越容易合成)、多样性、成功率(以上四个指标越高越好);
(3)运行时间。
3D-MCTS在运行时间与大多数方法持平的情况下,在八个指标中的七个上超越了现有方法。
表1 与其他方法对比
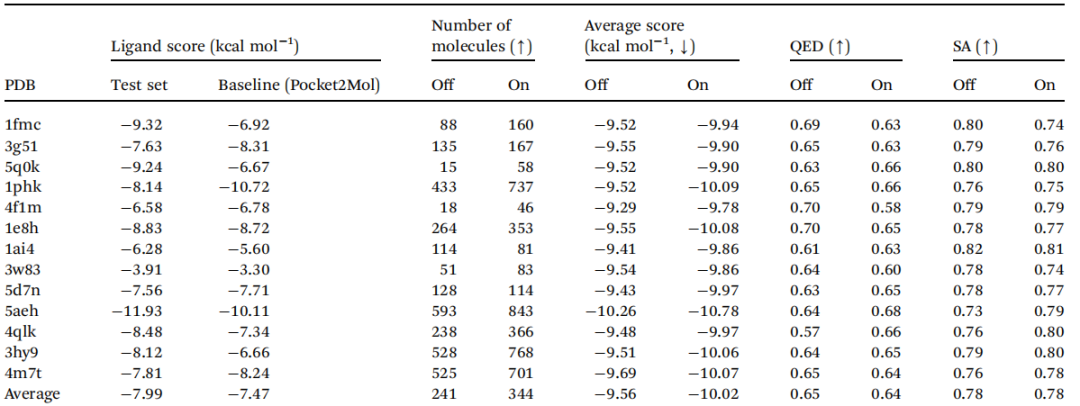
作者设计了模型消融实验,对比引入(on)或者不引入(off)药效团知识时的模型表现,并在不同的蛋白质上与基线模型(Pocket2Mol)进行对比,如表2所示。引入领域知识后,模型具有更低的对接分数(表现更好),有更多满足条件的分子可生成,而类药性和SA分数与之前持平。
表2 消融实验
总结
与其他方法相比,这些片段的使用使生成的分子具有最佳的内在化学合成性和药物相似性。为了优化搜索效率,该算法结合了多线程并行仿真和基于实时能量约束的剪枝策略。这使得它能够以固定的计算成本识别具有改进的目标结合亲和力的目标分子。
[1] Du et al. A flexible data-free framework for structure-based de novo drug design with reinforcement learning. Chem Sci. 2023
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢