目前,随着测序技术的不断发展,快速准确的蛋白质序列注释工具显得愈发重要。同时,随着各种高精度结构预测模型的出现,将结构信息同步到蛋白功能注释中也成为蛋白质组研究的新方向。2023年10月,来自纽约大学Data Science中心Kyunghyun Cho课题组的Tymor Hamamsy等人开发了通过蛋白质序列预测特征向量进而进行序列比对并完成序列注释的深度学习模型Protein-Vec [1],该工作目前在预印本平台BioRxiv上发布。该工具可针对蛋白结构-功能的7种指标进行特征向量的生成,并通过和预先生成的特征向量库进行余弦相似性比较的方式得到序列比对信息,最后完成序列注释。根据作者的测试结果,Protein-Vec在精度上超过传统的HMMER和BLASTp方法,同时也超过之前常用的基于深度学习的DeepFRI方法。——方法——
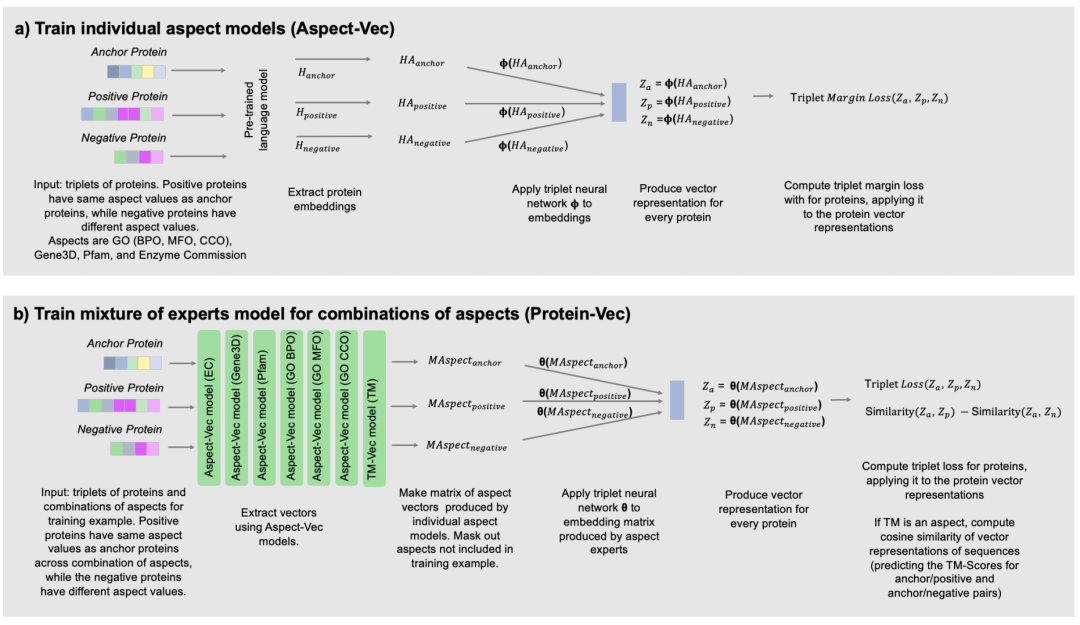
图1. Protein-Vec的模型架构
如图1所示,Protein-Vec基于对比学习训练 [2],主要分为两个部分,其中前半部分为分别预测7种蛋白质特征(EC编号、基因本体分子功能GO MFO、基因本体生物过程GO BPO、基因本体细胞组分GO CCO、Pfam、TM-Scores和Gene3D结构域注释)的特征向量的Aspect-Vec Embedding模型,后半部分是综合7种特征的Protein-Vec Embedding模型。在最前端的蛋白序列嵌入处,使用了之前发表的蛋白质序列预训练模型ProtTrans [3]。Protein-Vec是由ProtTrans、Aspect-Vec Embedding model,以及Protein-Vec Embedding model三者拼接而成。 Aspect-Vec Embedding模型的输入是蛋白质预训练模型的序列嵌入向量,输出是一个表示特征的512维隐空间向量。在Apsect-Vec训练完成之后,将7乘以512维的向量输入后面的Protein-Vec Embedding模型中,最后由Protein-Vec Embedding模型输出一个512维的向量作为蛋白质的整体特征向量。在训练过程中,作者提前对除了TM-scores之外的其他6种特征构建了“三元组”的训练样本,每个样本中包含对应当前序列的“锚定”蛋白,一个特征和“锚定”蛋白相近的正例蛋白以及一个负例蛋白。对于构建了三元组的特征,训练中的loss如下式所示:
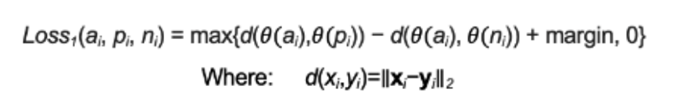
其中,a代表“锚定”样本的隐向量,p代表正例的隐向量,n代表负例的隐向量。θ代表神经网络模型。对于TM-scores,训练中的loss如下所示: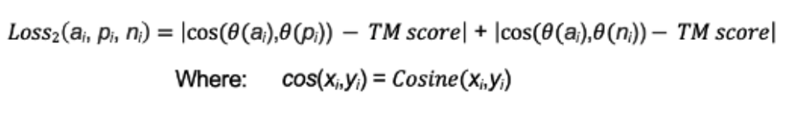
其中,a代表“锚定”样本的隐向量,p代表正例的隐向量,n代表负例的隐向量。θ代表神经网络模型。
对于Aspect-Vec Embedding模型而言,根据训练目标只使用Loss1或Loss2的其中一种;而对于Protein-Vec Embedding模型,在训练中使用对7种特征随机mask的策略,即输入的向量一部分是Aspect-Vec模型的输出向量,一部分是Mask向量。采取的loss为loss1和loss2的加和,若TM-scores部分被mask,则loss2设置为0。同时,对于训练数据的构建,作者也进行了一些特殊处理以使得模型具有更好的表现。整个数据集都是从Swiss-Prot中提取。对于锚定-正例-负例数据集的构建,作者特意在构建负例时考虑负例的难度,确保负例和正例的区分是一个“困难”任务。具体操作如下:1. 对于Pfam:正例被定义为与锚定蛋白质共享至少一个Pfam注释的蛋白质,而负例则采样了具有不同Pfam注释但共享至少一个Pfam家族的蛋白质。2. 对于Gene3D:正例被定义为与锚定蛋白质共享至少一个Gene3D注释的蛋白质,而对于负例,作者使用Gene3D本体来采样与锚定蛋白质具有相同体系结构或相同拓扑的负例3. 对于EC编号:已知EC分类共有4个分级。对于正例,使用共享至少一个EC注释的蛋白质(在所有4个级别上);对于负例,采样不与锚定蛋白质共享任何EC注释但具有与EC层次结构的前2级或前3级一致的EC注释的蛋白质。4. 对于GO:使用GOGO工具,将阈值设定为0.65,分别对生物过程、细胞组分和分子功能组件三种特征生成三元组数据5. 对于TM-scores:使用TM-align工具。最终数据包含约4.5亿个“三元组”样本。划分的测试数据集一种有3组,其中一组是按时间划分,即2022年5月之后提交到Swiss-Prot上的样本,另外两组则是序列同源性小于50%和20%的测试数据集。完成训练后,Protein-Vec的使用过程如下图2所示: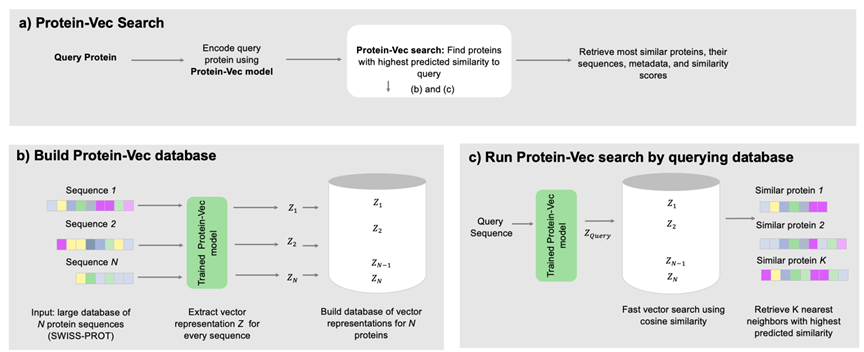
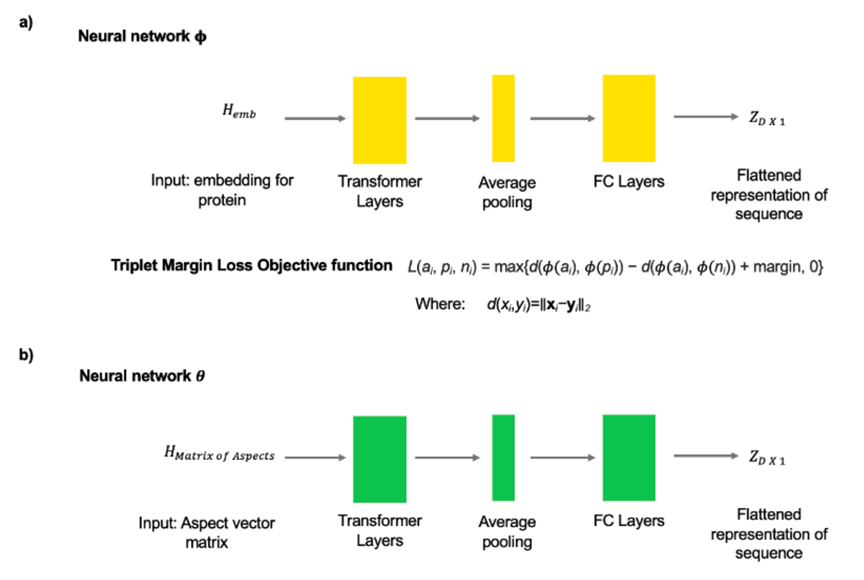
图3. Aspect-Vec Embedding和Protein-Vec Embedding的网络架构——结果——
作者随后对Protein-Vec的注释效果进行了测试。EC分类预测如下表1所示:
可见,Aspect-Vec模型和Protein-Vec模型都比之前的CLEAN分类更加精确,同时Protein-Vec的表现超过Aspect-Vec许多,说明将其他信息(包括结构信息)整合到一起对酶分类任务也有长足提升。
表2. Pfam比对与之前的传统比对方法及之前的深度学习方法比较可见,Aspect-Vec模型也具有更好的效果。
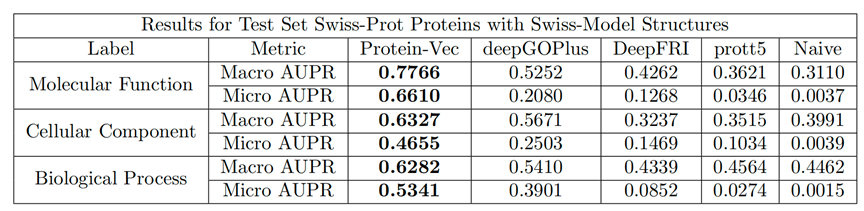
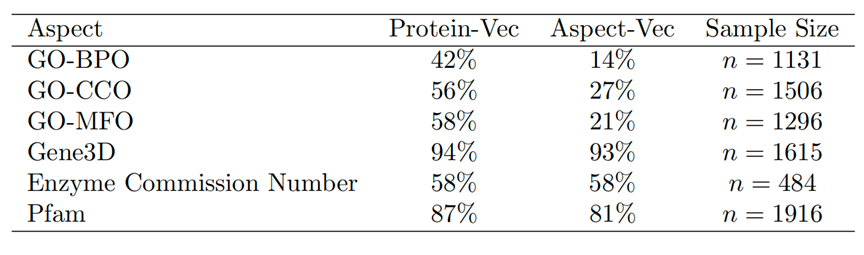
表4. Protein-Vec与Aspect-Vec在各指标上的Recall值可见,Protein-Vec在各种特征的表现上都高于单独的“专家”模型Aspect-Vec,这说明多特征的整合确实具有一定效果。 同时,作者还发现Protein-Vec在对于低同源的测试集表现弱于高同源性的测试集,如下表5-6所示: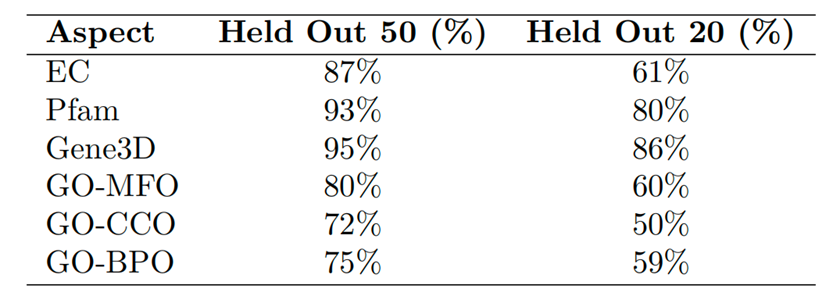
表5. 在50%同源性测试集和20%同源性测试集上的recall表现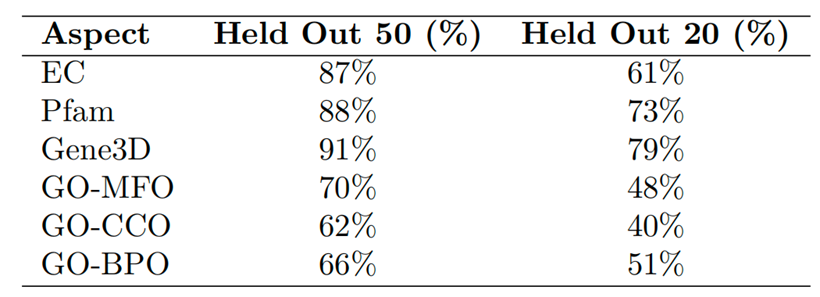
表6. 在50%同源性测试集和20%同源性测试集上的accuracy表现可见,作为基于深度学习模型的Protein-Vec,还是存在深度学习模型的泛化能力通病。不过作者表示即使是在低于20%序列同源性的测试集上,Protein-Vec相比于之前的方法也具有相当的竞争力。——小结——
总的来说,Protein-Vec提供了一种将蛋白序列直接表示成特征向量的工具,并且在作者的benchmark上表现很好。在当下这个测序数据爆发的时代大有可为。 当然,Protein-Vec也存在很多需要改进的问题,例如对于某些低同源性序列的泛化能力,以及关于多标签的预测。比如,对于GO注释,很多蛋白其实有多个标签,而在Protein-Vec中,则只预测最邻近的标签。另外,作者在文末表示Protein-Vec对于局部相似性预测很差,但并未给出具体的图表数据。[1] Tymor Hamamsy, Meet Barot, et al. bioRxiv 2023.11.26.568742; doi: https://doi.org/10.1101/2023.11.26.568742[2] Gregory Koch, Richard Zemel, Ruslan Salakhutdinov, et al. Siamese neural networks for one-shot image recognition. In ICML deep learning workshop, volume 2, page 0. Lille, 2015.[3] Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, Debsindhu Bhowmik, and Burkhard Rost. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence, 44(10):7112–7127, 2022.
点击左下角的"阅读原文"即可查看原文章。
内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢