
今天为大家介绍的是来自Pietro Sormanni和Michele Vendruscolo团队的一篇论文。非天然氨基酸能扩展化学空间,定制肽类药物的功能、半衰期和其他关键属性。尽管含有修饰氨基酸(如经过翻译后修饰的残基)的化学空间极大,但实验测量含修饰氨基酸肽的开发性质既昂贵又耗时。为了通过计算方法促进开发性项目,作者提出了CamSol-PTM方法。这种方法能快速、可靠地基于序列预测含修饰氨基酸肽在室温下水溶液中的内在溶解度。

与小分子药物相比,肽类药物具有多种优势:通常毒性低,不易在组织中积累,因此既安全又高效。肽类药物多样、高效、易于合成,并且由于其相对较大的尺寸,具有比小分子药物更高的特异性。然而,肽类药物候选物也存在一些问题。它们通常具有低口服生物利用度和短半衰期,这是由于高清除率和低代谢稳定性造成的。此外,肽类药物可能具有较差的膜透过性。
自然界中的例子表明,内源性肽和蛋白质的特性可以通过翻译后修饰(PTM)进行改变。典型的PTM包括用于信号转导和能量代谢的磷酸化,以及用于调节的乙酰化和糖基化。其他常见的修饰包括酰胺化等等。在实际治疗应用中,研究人员通过PTM改善肽性质,如增加生物活性和改善代谢稳定性,增加结合亲和力、半衰期以及通过脂肪化和酰化改善组织渗透能力等等。通过采用扩展PTM范围的策略,改造氨基酸(mAAs)的使用在生物技术和药物开发中变得突出。
改造氨基酸可以包含新的功能团,改变肽的主链或末端结构。mAAs的效果多样,可以针对生物制品固有的特定问题进行修饰。肽药物开发的一个主要问题是被蛋白酶和肽酶识别,可以通过改变主链结构来减少这一问题,例如加入酰胺键模拟物、D-异构体、β-氨基酸、改变末端或四取代氨基酸。这些模拟物还倾向于增加生物利用度,降低构象灵活性。类似效果也可以通过N-烷基化、加入异丁氨酸、其他限制性氨基酸或通过环化来实现。后者和添加空间大的基团还可以减少T细胞识别。通过糖基化可以改善生物利用度和稳定性,增强蛋白质-蛋白质相互作用,利用细胞表面的葡萄糖转运蛋白提高细胞渗透性。通过增加疏水性也可以改善渗透性,可以通过甲基化、脂肪化、加入氟化残基或修改末端残基来实现。
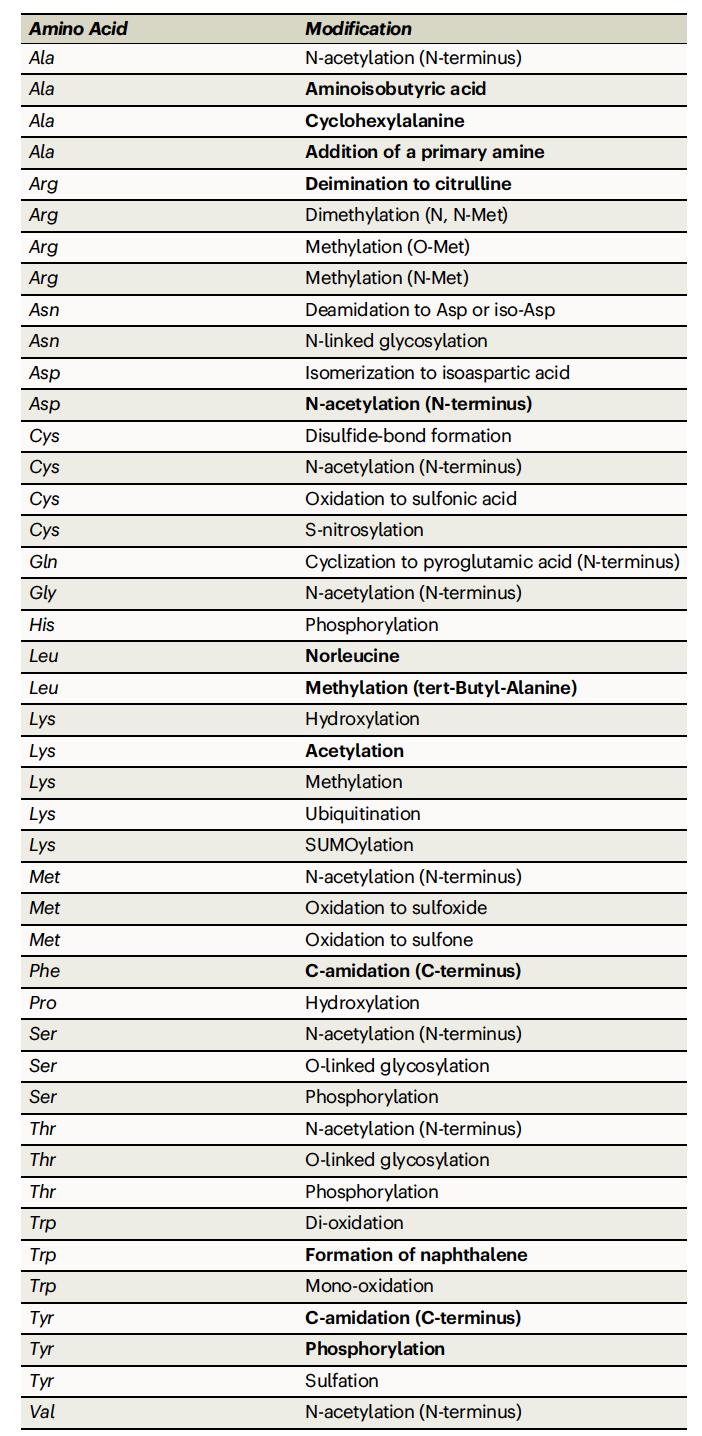
表 1
商业供应商目前提供了数百种可合成的改造氨基酸(mAAs),这些mAAs可以合成到肽中。然而,用于表征肽的实验方法往往既耗材料又耗时。如最先进的溶解度测量方法需要大量的材料,且通常不适用于成千上万候选物的筛选。因此,开发能预测含有mAAs的肽和蛋白质的内在溶解度和聚集倾向的计算方法将非常有益。通过在开发流程中整合快速且廉价的计算机筛选,可以避免或大大减少费力的溶解度测量。尽管已有几种准确的蛋白质和肽溶解度预测器,以及对单个氨基酸的预测器,但目前还没有基于序列的方法可以方便地处理非天然氨基酸。为了填补这一空白,作者利用CamSol框架预测内在溶解度,并开发了CamSol-PTM方法。该方法可以处理含有与标准氨基酸大小相似的mAAs的肽。CamSol-PTM能够评估任何类型的小型非典型氨基酸对肽在室温下水溶液中内在溶解度的影响。
模型部分
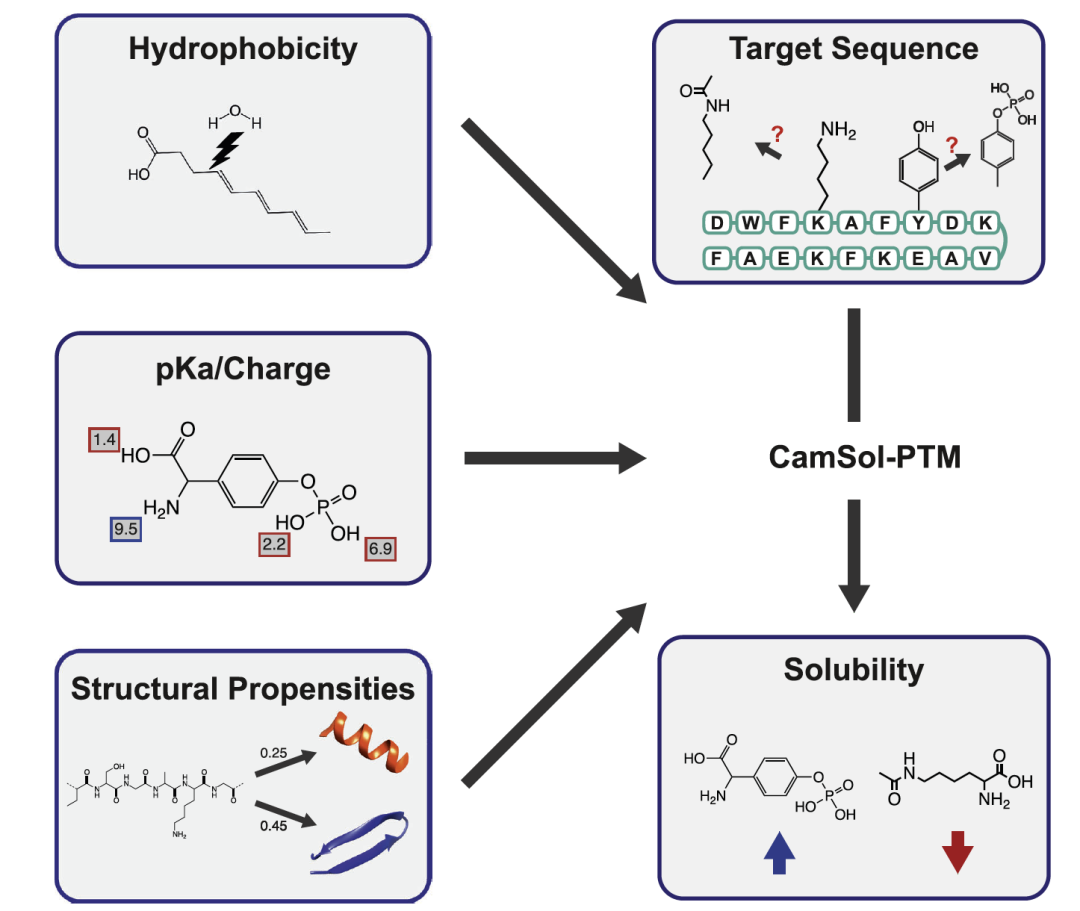
图 1
在这项工作中作者采用了CamSol框架,该框架原本用于准确预测蛋白质的内在溶解度,现在则用于引入一种预测mAAs对肽溶解度影响的方法。原始的CamSol方法通过结合20种标准氨基酸的疏水性、电荷、α-螺旋和β-折叠倾向的数值来预测蛋白质的内在溶解度。为了将这些数值扩展到不同的mAAs,需要这些mAAs的物理化学性质信息。由于作者的目标是估算含mAAs肽的内在溶解度,而不是进行广泛的实验研究,因此作者建了一个流程,其中mAAs的物理化学性质是通过计算方法预测的。在实际操作中,作者使用了pIChemiSt软件来计算修饰侧链的pKa值。肽的亲疏水性预测,作者采用ALOGPS和XLOGP3的集成方法。结构倾向预测也是由多个常见的由rdkit计算的物化属性构建的预测器预测。
数据来源
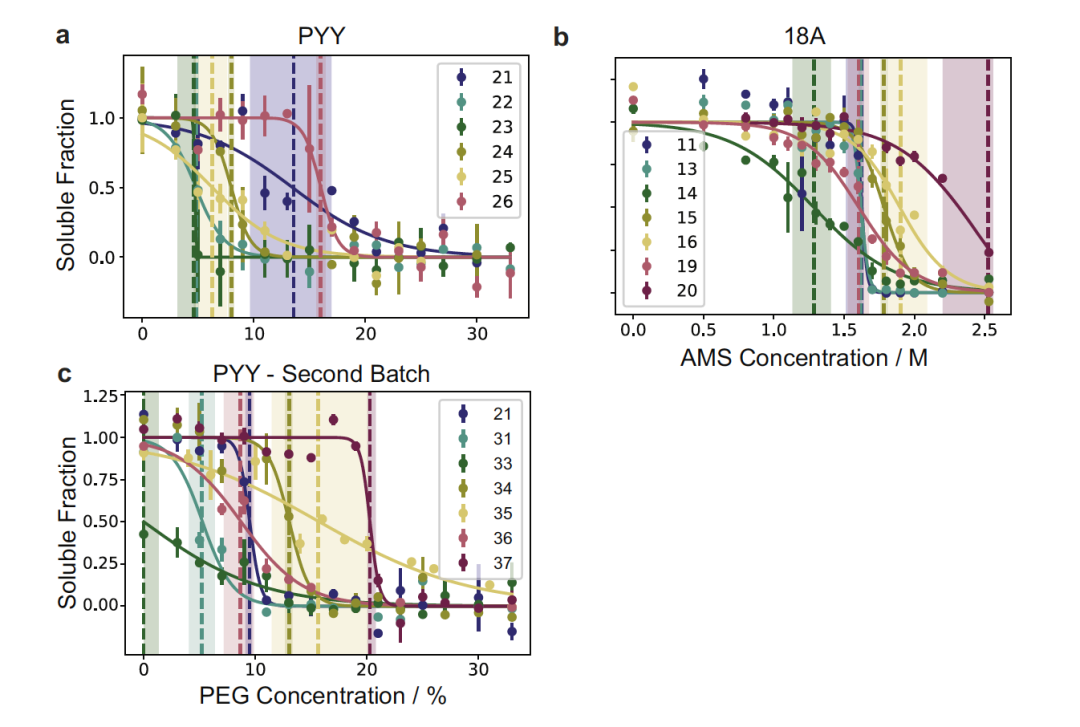
图 2
作者对胰高血糖素样肽-1(GLP-1)、酪氨酸酪氨酸(PYY)和载脂蛋白A的衍生物(18A)这三种肽进行了氨基酸改造(mAAs)的修改,以研究这些改造对肽的结构和功能的影响。对于GLP-1,由于它由两个α-螺旋组成,中间通过连接区分隔,作者选择了连接区的第一个丙氨酸(残基24)作为单点修饰的起始点。对于双点修饰,他们排除了一些在结合中起着关键作用的残基,如7His、8Ala、9Glu等,以保持GLP-1的功能性。对于PYY,由于它在N端含有富含脯氨酸的α-螺旋,与构成分子其余部分的α-螺旋形成氢键,作者选择了脯氨酸丰富区域的一个丙氨酸进行单点修饰。在双点修饰中,他们排除了所有脯氨酸和形成氢键的残基,如R、H、K、D、E、N、Q等。而18A具有两面性的特点,对于单点修饰,作者选择了位于两侧边缘的位置10的丙氨酸。在双点修饰中,他们确保了亲水残基(D、E、K)只被亲水性修饰(CIT、AIB)替换,而疏水残基(W、F、A、V)只被疏水性mAAs(CHA、NAC、NLE)替换。实验测定的部分结果见图2。
实验结果
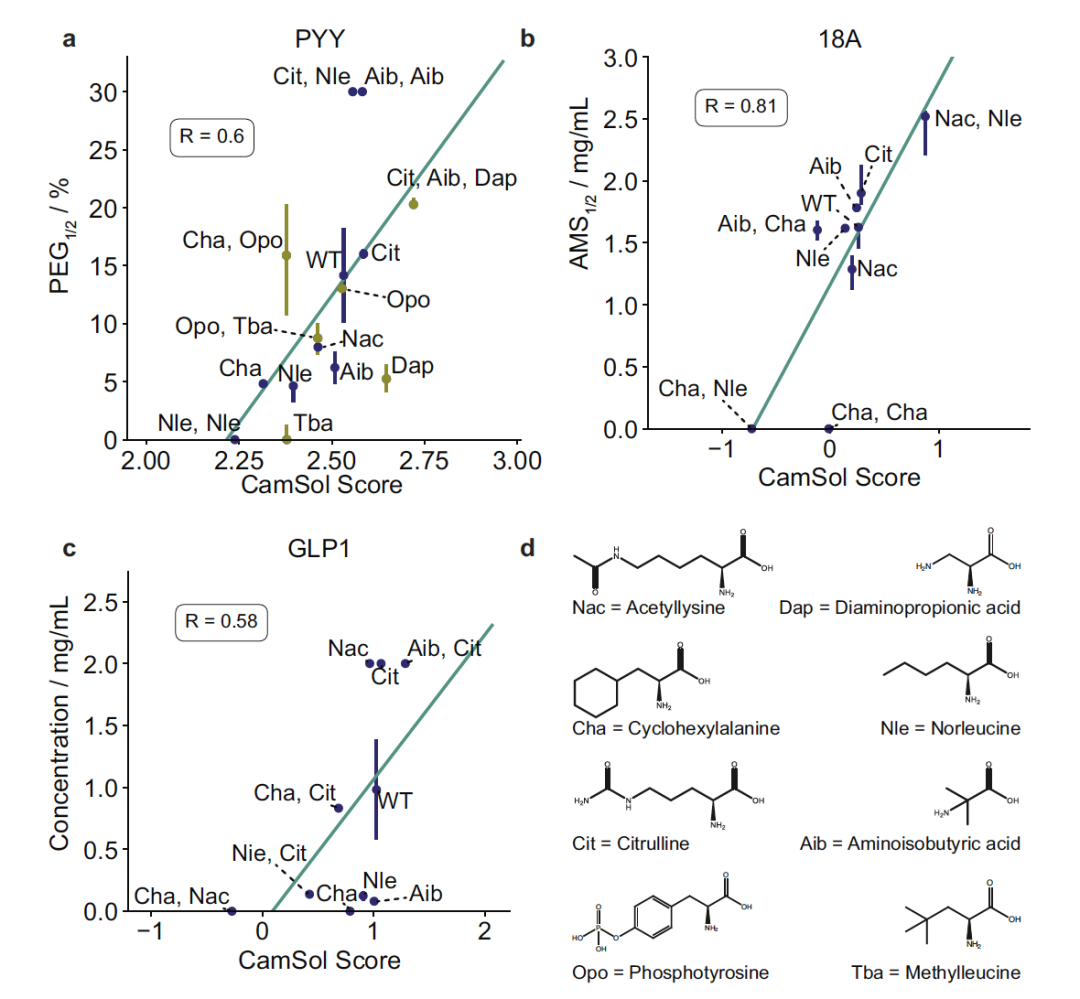
图 3
通过将计算预测与实验数据进行比较,研究发现两组数据之间存在高度相关性。酪氨酸酪氨酸(PYY)变体的皮尔逊相关系数为0.78,18A变体为0.81,胰高血糖素样肽-1(GLP-1)变体为0.58(见图3)。为了确认这些发现不是巧合,作者设计了一组新的PYY变体,包含四种新的mAAs,并测量了它们的溶解度(见图2c)。这些结果在图3a中以赭色显示。PYY变体的整体皮尔逊相关系数为0.6。
由于实验验证的结果令人鼓舞,作者开始推广计算方法,以拓宽其适用性,覆盖更多mAAs类型。他们建立了一个网站,供学术用户免费使用这种方法。他们通过用RDKit中的Crippen工具替换亲疏水性预测器,自动化了添加新mAAs的过程。如果用户想预测包含尚未实施的非典型氨基酸的肽的溶解度,只需提供SMILES代码。提供这些信息后,网站将自动计算该mAAs所需的属性,以便用户在预测中包括它。
网站地址:
https://wwwcohsoftware.ch.cam.ac.uk/index.php/camsolptm
编译 | 曾全晨
审稿 | 王建民
参考资料
Oeller, M., Kang, R. J., Bolt, H. L., Gomes dos Santos, A. L., Weinmann, A. L., Nikitidis, A., ... & Vendruscolo, M. (2023). Sequence-based prediction of the intrinsic solubility of peptides containing non-natural amino acids. Nature Communications, 14(1), 7475.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢