
今天为大家介绍的是来自William A. Donald团队的一篇论文。论文介绍了一种基于机器学习(ML)的方法,用于处理代谢组数据,旨在早期诊断疾病。虽然机器学习与代谢组学的结合提供了早期诊断疾病的机会,但由于解释疾病预测模型的挑战以及分析大量相关且“嘈杂”的化学特征的困难,这种方法的准确性和获取的信息量可能受到限制。在这项研究中,研究团队报告了一种可解释的神经网络框架,它可以准确地预测疾病并在不需要预先选择特征的情况下,使用完整的代谢组数据集识别重要的生物标志物。

帕金森病的发病率正在迅速增长,它通常可以根据运动症状的临床标准来诊断,这些症状包括运动迟缓、静息性震颤和僵硬。然而,一些非典型的非运动症状,如睡眠障碍、便秘、冷漠和嗅觉丧失,可能在临床相关症状出现前几年甚至几十年就已存在。此外,对于表现出类似帕金森病症状的患者,目前诊断的过程经常是不确定的。使用与代谢过程相关的生物标志物在理解、诊断和监测疾病应用方面非常广泛。这些代谢物通常来自于血浆和血清等已建立的样本,通过质谱(MS)分析可对成千上万的代谢物进行痕量分析。利用质谱分析,在帕金森诊断前15年,预帕金森患者的血浆代谢物与未患帕金森的健康对照组相比已有所不同。这些结果表明,帕金森可能可以使用代谢物生物标志物更早地诊断,尤其是如果这样的分析能够提高诊断准确性,并在大型队列研究中得到验证。
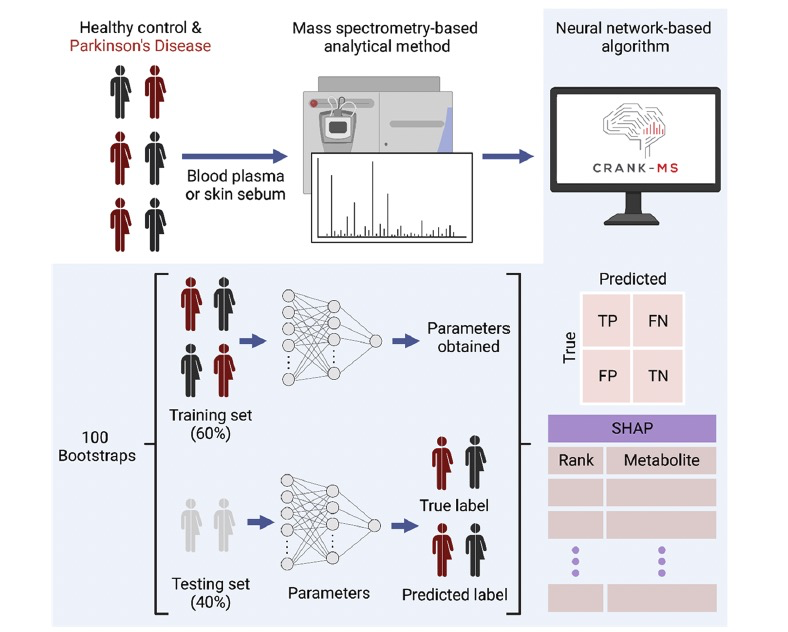
图 1
传统上,由于代谢组数据集包含高度相关且“嘈杂”的化学特征,这可能导致模型过度训练并降低诊断性能,因此常常基于较小的特征子集构建模型,这些特征子集通常通过传统统计方法确定。例如,Gonzalez-Riano等人使用支持向量机(SVM)模型,并预先选定20个生物标志物来诊断血浆样本中的预帕金森与健康对照组。Sinclair等人使用最小二乘判别分析(PLS-DA)和预先选定的15和26个生物标志物分别诊断未用药和用药的帕金森患者与健康对照组。
然而,由于代谢物的丰度通常相互关联,并且可能与其他代谢物的丰度非线性依赖,像SVM和PLS-DA这样的ML方法可能会“错过”代谢组数据集中的一些关键特征。高级ML方法如神经网络(NN)特别适合处理大量相关数据,并构建包含非线性效应的数据集的模型。但使用NN等方法对基于代谢组数据的复杂混合物进行分类时,一个根本问题是产生的预测模型通常被视为不可解释的“黑箱”,无法直接用于揭示机理信息。
最近,一种名为Shapley Additive exPlanations(SHAP)的新方法被开发出来,用于通过事后计算单个特征对模型准确预测性能的贡献来“解释”ML模型。然而,SHAP尚未用于代谢组数据集的分析,因为解释ML模型的方法只是最近才被开发出来,而且使用所有化学特征可能导致预测模型的过度训练。理想情况下,整个代谢组数据集应该被包含在ML模型中,以便SHAP能够识别推动模型预测的关键代谢物。这里,作者报告了一个基于可解释神经网络的框架,用于分析基于非靶向质谱方法生成的数据集,名为“CRANK-MS”(基于神经网络的质谱生成知识的分类和排名分析)(图1)。
数据来源

表 1
研究采用了两个帕金森病代谢组学研究的数据集。首先,西班牙欧洲营养与癌症前瞻性研究(EPIC)研究涉及从后来发展为帕金森病的受试者和未发展为帕金森病的受试者中采集的血浆样本的代谢组数据,时间跨度长达15年,参与者总数为78人。EPIC研究中的血浆样本通过四种不同的仪器方法进行分析,包括气相色谱-质谱(GC-MS)、毛细管电泳-质谱(CE-MS)以及在正离子模式和负离子模式下的液相色谱-质谱(LC-MS)。第二个数据集来源于NHS研究,涉及对未服用药物和已服用药物的帕金森病患者以及健康对照组(共274人)的皮肤皮脂进行LC-MS(正离子模式)分析(表1)。
方法
研究团队使用代谢组数据作为输入,利用六种监督学习框架来区分帕金森病患者和健康人群。这些机器学习方法包括随机森林(RF)、梯度提升(XGB)、线性判别分析(LDA)、逻辑回归(LR)和支持向量机(SVM),它们都是使用scikit-learn包)编写的。算法和SHAP(Shapley Additive exPlanations)分析是在Python中实现的。此外,使用PyTorch写了带有多层感知器的神经网络(NN)。
实验部分
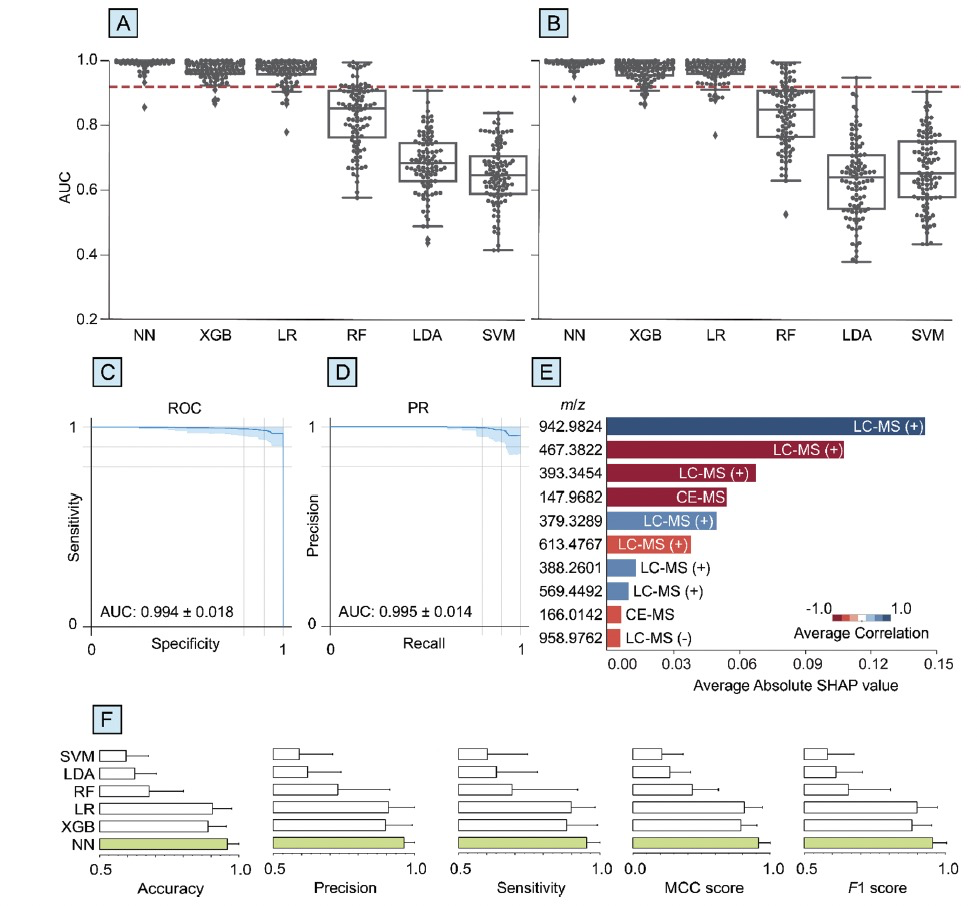
图 2
首先,六种机器学习算法的整体诊断性能是基于血浆代谢物的复合数据集进行评估的,该数据集包括四种分析方法检测到的代谢物。在所有评估指标上,神经网络的诊断性能都高于其他框架(图2)。具体来说,在使用神经网络进行帕金森与健康的二分类时, ROCAUC为0.994 ± 0.018,而PRAUC为0.995 ± 0.014。梯度提升和逻辑回归的表现与神经网络相近,但稍低。相比之下,随机森林、支持向量机和线性判别分析的表现较低。
SHAP分析被用来识别对帕金森预测贡献最大的代谢物和相应的质谱分析方法。在血浆的复合代谢组数据集中,前六名代谢物中有五个是使用液相色谱-质谱(LC-MS)正离子模式检测到的。为了进一步验证这些化学特征在预测帕金森方面的贡献,所有六种机器学习算法都应用于仅包括LC-MS(+)数据的数据集,而不包括LC-MS(−)、GC-MS和CE-MS数据集。与复合数据集相似,使用NN和LC-MS(+)数据进行PD的二分类在所有性能指标上都是最高的。这些结果表明,LC-MS(+)数据集在所有六种机器学习方法中的诊断性能优于其他三种质谱分析方法。使用神经网络得出的诊断准确性高于其他五种机器学习方法,而且LC-MS(+)数据集的化学特征数量(509个)远少于复合数据集(1430个)。简而言之,这一部分展示了神经网络在处理帕金森诊断中的代谢组数据方面的强大性能,尤其是在处理大量相关数据和建立包含非线性效应的模型时。
随后作者讨论了在诊断性能方面,将整个代谢组数据集纳入模型的影响。与仅使用预先筛选的化学特征训练模型不同,神经网络模型包括了数据集中的所有代谢物或特征。例如,在Gonzalez-Riano等人的研究中,首先对帕金森的生物标志物进行重要性筛选,然后在最终的诊断模型中使用这些生物标志物的一个小子集(最多20个)。在这项之前的研究中,使用20个特征的线性支持向量机模型在复合数据集中获得的最高ROC(AUC)值为0.919。然而,在当前研究中,应用于复合数据集的类似模型(不进行特征选择)的AUC(ROC)仅为0.647 ± 0.093。相比之下,使用NN对复合数据集中的所有特征进行处理,得到的AUC(ROC)为0.994 ± 0.018。这些结果与一些知名的机器学习模型在包含许多“嘈杂”特征的大型数据集中预测性能相对较低的情况一致。
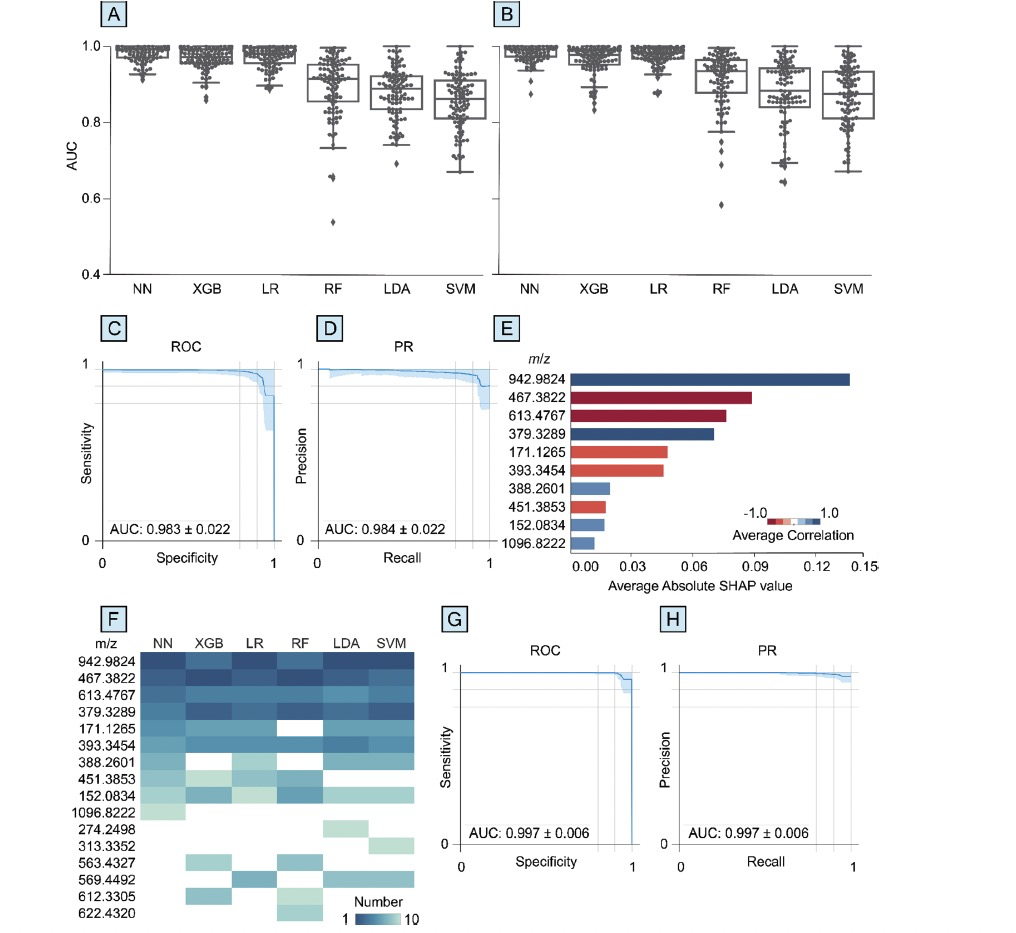
图 3
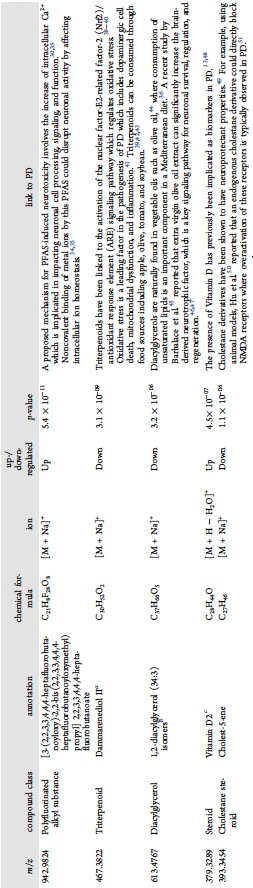
表 2
通过SHAP分析,研究人员能够识别出对帕金森准确预测最有贡献的代谢物,这些代谢物更可能是基性的,容易通过阳离子加成被电离,而不是酸性的。这一发现是基于使用液相色谱-质谱(LC-MS)正离子模式比负离子模式获得了更高的PD诊断性能(图3),以及每种方法测量的代谢物数量大致相同(约510至530个)。在血浆的LC-MS(+)数据上进行的SHAP分析显示,前六个最高得分的代谢物在所有六种机器学习算法中都是一致的。这些检测到的代谢物与之前聚焦于线性支持向量机模型的研究确定的代谢物不同,这可能是由于使用基于核的方法和神经网络之间的差异。对帕金森准确预测贡献最大的五种代谢物可以作为疾病状态的潜在指标,并已进行注释(表2)。这五种注释的离子包括多氟烷基物质(PFAS)、三萜类、胆甾烷类固醇、二酰基甘油和维生素D类固醇,这些成分可能是内源性或外源性的,且之前的文献已将它们与帕金森联系起来。
例如,质荷比为942.9824的离子具有最高的SHAP值,可能对应于钠化的PFAS(DTXSID70325550)。PFAS及其氧化和水解产物在帕金森病例中的水平高于健康对照组。PFAS化合物在环境和人类血液中无处不在,因为它们具有生物累积能力、化学稳定性和广泛用于工业和消费品,如塑料、不粘锅和食品包装。DTXSID70325550是一种PFAS化合物,目前列在美国环境保护署CompTox化学品数据库中,它似乎容易与Na+、K+、Ca2+、Cu2+和Zn2+结合。因此,这种化合物可能通过影响细胞内离子稳态来干扰神经活动。PFAS诱导的神经毒性的潜在机制之一涉及细胞内Ca2+的增加,这可能影响神经细胞的处理、信号传递和功能。虽然需要进一步的体外和体内研究来调查DTXSID70325550对神经细胞功能的影响,但这些数据表明血浆中特定PFAS化合物的升高水平可能是帕金森的早期指标。总的来说,这些结果进一步支持SHAP分析在识别帕金森潜在生物标志物方面的用途,这些生物标志物最初未通过统计方法发现。
编译 | 曾全晨
审稿 | 王建民
参考资料
Zhang, J. D., Xue, C., Kolachalama, V. B., & Donald, W. A. (2023). Interpretable machine learning on metabolomics data reveals biomarkers for parkinson’s disease. ACS Central Science.
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢