
今天为大家介绍的是来自Michiaki Hamada和Hirohide Saito团队的一篇论文。RNA工程在生物技术和医学领域有巨大潜力。尽管RNA工程非常重要,但目前仍缺乏一个多功能的自动化设计功能RNA的平台。因此,作者们提出了一种名为RfamGen的深度生成模型。这个模型通过明确结合序列对齐二级结构信息,以数据高效的方式设计RNA家族序列。RfamGen能够通过从一个语义丰富且连续的表示中采样点来生成新颖且功能性的RNA家族序列。作者们通过多种RNA家族的实验,证明了RfamGen的多功能性。

在功能性RNA设计的计算尝试中,主要研究了RNA逆向折叠的概念,这是一种寻找能够折叠成给定二级结构的序列设计方法,由RNA二级结构预测和离散优化算法指导。然而,由于RNA的功能性不仅仅由结构特征,所以仍需以临时方式选择设计的序列。此外,由于RNA逆向折叠的灵活性和通用性不足,它在生成具有期望功能的可接受变异的序列方面存在困难,其准确性受RNA二级结构预测和优化算法的准确性限制。不同于RNA逆向折叠,协变模型(CM)是一种用于RNA序列对齐和共识二级结构的统计框架,可以在不依赖RNA二级结构预测的情况下定量评估序列和结构的变异。由于其灵活性,CM已经成为几十年来RNA同源性搜索的黄金标准,将大多数功能性RNA物种分类为数千个“RNA家族”。尽管CM以前没有用于功能性RNA设计,但它可能为避开RNA逆向折叠相关的技术难题提供了一个有希望的框架。
此外,通过离散算法探索指数级庞大的序列空间所带来的优化难题,可以通过序列表示来缓解。序列表示是一种基于机器学习的方法,将离散序列嵌入到连续空间中。为了高效设计具有期望属性的序列,学习具有连续性的有意义的表示非常重要。最近,深度生成模型因其学习语义丰富的序列表示而受到了广泛关注。特别是,内部表示为“潜在空间”的深度生成模型,如变分自编码器(VAE),能够生成模仿现有数据,同时将原始数据嵌入到潜在空间中。因为潜在空间允许数据的连续内插,VAE使得可以通过连续优化生成具有期望属性的序列。在这项研究中,作者提出了RNA家族序列生成器(RfamGen),这是一个用于功能性RNA设计的深度生成模型。RfamGen利用VAE和CM生成RNA家族的人工序列,同时提供序列的语义上有意义的表示。作者通过体内和体外方法证明了RfamGen对多种RNA家族的广泛适用性。
RfamGen模型
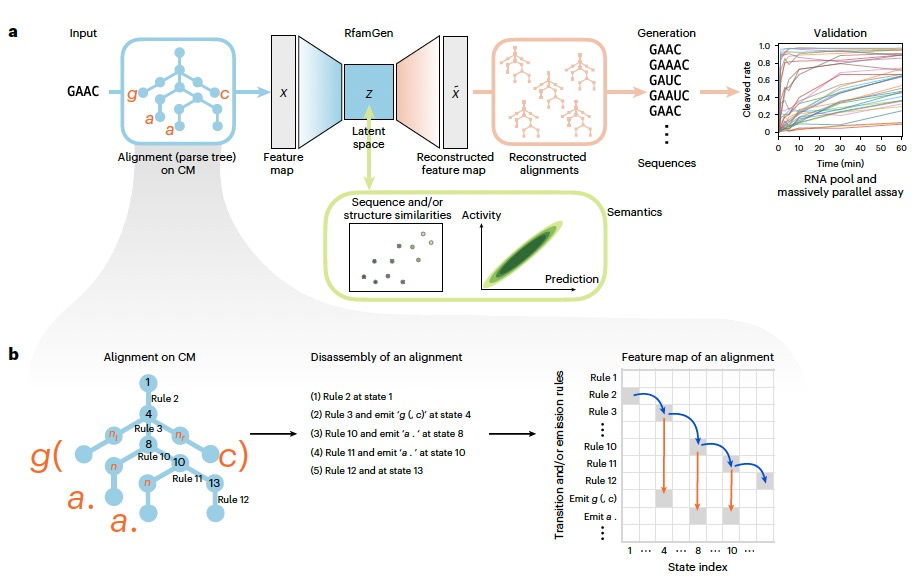
图 1
RfamGen是一种利用协变模型(CM)架构的变分自编码器(VAE),用以提高RNA家族序列生成的效率。CM是一种基于树结构的隐马尔可夫模型,用于在共识二级结构的约束下形式化多序列比对(MSA)。在这个框架中,MSA中的每个序列都被对齐并表示为在CM上的一条路径(图1b)。由于CM明确描述了MSA中变异的结构约束,通过VAE架构,RfamGen被训练来恢复模仿自然序列的对齐,同时将它们嵌入到潜在空间中。为此,作者将对齐到CM的特征向量化,然后旨在使用VAE生成CM上的路径,而不是直接生成序列。作者将路径分解为CM中的转移和生成规则,并将它们转换为one-hot表示,接着进行进一步处理以稳定学习过程。在采样过程中,模型通过重建CM并通过从重建的CM中取最大值(argmax)的参数来生成序列。
RfamGen是一个高效生成器
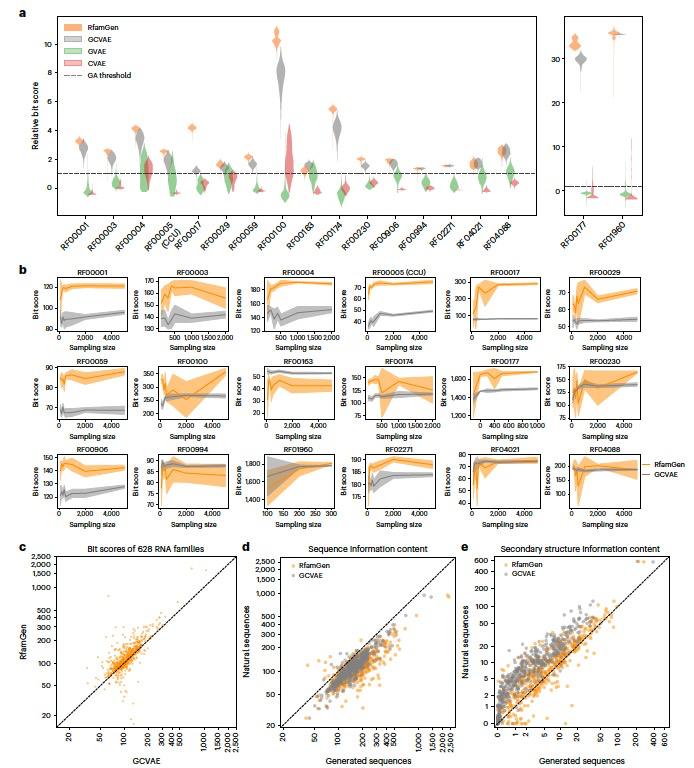
图 2
为了检验显式使用协变模型(CM)在生成能力上的影响,作者首先将RfamGen与三种未考虑二级结构和/或序列对齐信息的消融模型(ablation models)进行了比较。这些模型分别是:只包含序列对齐信息的GCVAE(gapped character VAE)、只包含二级结构信息的GVAE(grammar VAE)以及两者都不包含的CVAE(character VAE)。作者通过比较这些模型生成的序列质量(使用“比特分数”来衡量,根据序列对Rfam数据库中的CM进行对齐来计算),来评估它们的性能。研究结果显示,RfamGen和GCVAE在序列生成方面的能力较强,其次是GVAE和CVAE,尽管RfamGen在大多数情况下表现最佳(图2a,b)。这些结果表明,加入序列对齐信息显著提升了性能,而二级结构信息则适度改善了性能。接下来,作者通过对不同数据规模进行欠采样,评估了RfamGen和GCVAE的有效性和稳健性。RfamGen在广泛的采样规模下保持了高生成能力,对于大多数RNA家族而言,仅用约500个输入序列就能达到接近顶峰的性能。最后,研究者们使用至少有100个序列的628个RNA家族的完整对齐数据对RfamGen进行了训练,并与GCVAE进行了比较以确认其应用范围。结果显示,RfamGen生成的序列比特分数高于GCVAE,从而证明了RfamGen在生成RNA家族序列方面的优越性。
此外,研究还调查了这两个模型在学习自然序列信息内容方面的能力。研究者们计算了自然和生成序列的序列信息熵和基于CM共识二级结构的成对生成概率的互信息。尽管RfamGen在一些RNA家族的序列信息内容学习方面稍显不稳定,但在再现二级结构信息内容方面显著优于GCVAE。
RfamGen学习了一个语义丰富的潜在空间
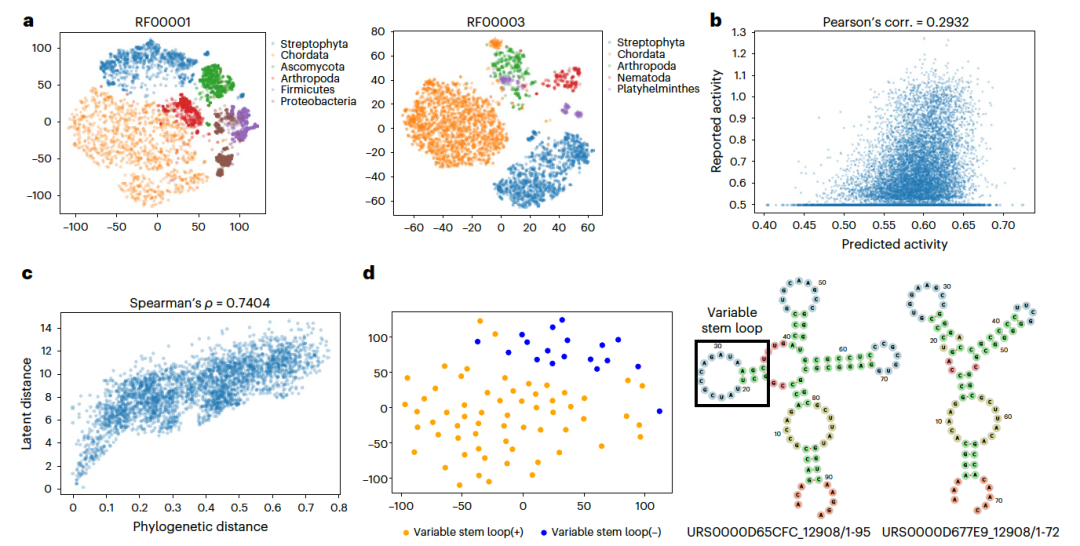
图 3
RfamGen学习了一个语义丰富的潜在空间,这对序列设计至关重要,因为潜在空间需要包含丰富的语义信息。为了检验RfamGen的潜在空间是否包含有意义的序列表示,作者进行了分析。他们利用t-SNE方法,对RfamGen的16维潜在空间进行了三维投影,图3a。结果显示,RfamGen的潜在空间展现出了基于系统发生学的分离的簇。在其他RNA家族的分析中也观察到了类似的趋势。此外,作者还使用了tRNA的深度突变扫描(DMS)工具,来探究RfamGen如何将序列变体嵌入到潜在空间中。他们进行了线性回归分析,以近似潜在空间上的活性景观,并发现两者之间存在中等程度的相关性(r = 0.2932,图3b)。这表明潜在空间线性地表示了tRNA的活性。
RfamGen用于生成多种RNA家族的活性序列
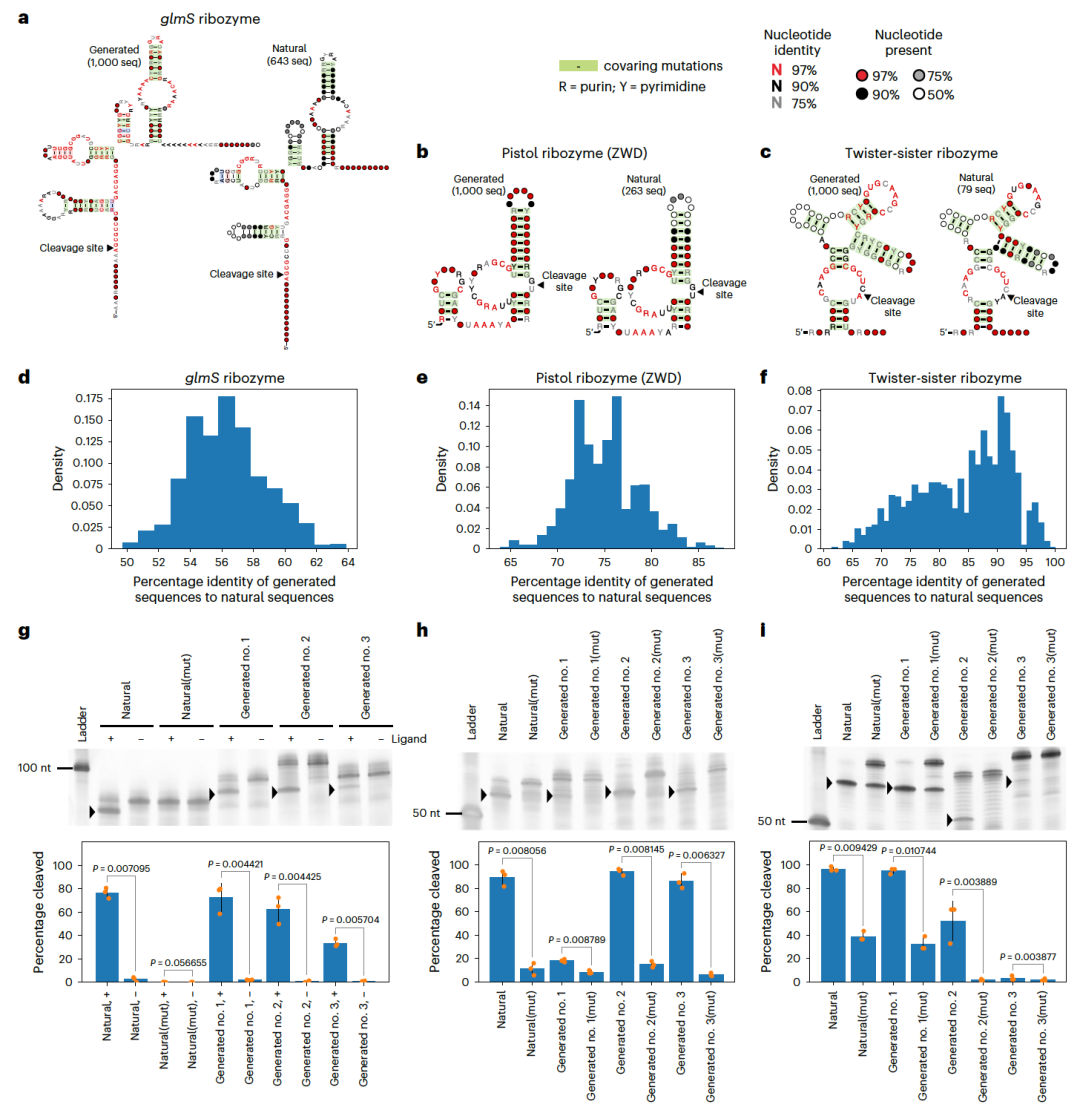
图 4
在RNA工程领域,自切割核糖酶是最广泛研究的功能性RNA之一。作者评估了RfamGen生成功能性序列的适用性,使用了来自Zasha Weinberg数据库(ZWD)和Rfam数据库的多种自切割核糖酶,这些核糖酶的序列数量相对较少(约100至800个)。由RfamGen生成的序列展示出与自然序列相似的序列和结构特征,但序列同一性有所不同。虽然RfamGen限制了核糖酶进化上保守的二级结构区域,但它能产生具有高序列多样性的RNA家族序列。为了评估生成序列的功能,研究者随机合成了每种核糖酶的三个生成序列,并设计了相应的突变体,这些突变体据报道活性降低。与此同时,他们也对一个自然序列进行了测试。通过电泳确认了序列的切割活性。与RfamGen生成的序列不同,研究者发现通过CM随机采样生成的序列没有活性。CM的随机采样在理论上具有类似的序列和/或二级结构偏好,但没有诸如更高结构约束或碱基配对堆叠等多体相关性。因此,研究结果支持RfamGen学习数据中的多体相关性的能力。
RfamGen用于预测多种RNA家族变体的效果
在RNA工程中,经常需要设计具有调整活性的序列。为此,研究人员已经进行了高通量实验,以筛选出最佳活性的变体,用于工程应用。另一方面,在蛋白质工程中,根据经验知道,可以通过使用自然序列训练的生成模型的生成概率来预测活性。在之前的一些研究中,tRNA也有类似的观察结果。然而,目前还不清楚这种方法是否普遍适用于多种RNA家族。因此,作者扩展了这一分析到更多的RNA家族。他们通过手动整理以前DMS研究中的序列变体活性数据。接着,他们检验了是否可以用与蛋白质相同的策略预测序列变体的活性。使用整理后的数据集,作者比较了序列变体的报告活性和RfamGen的损失函数。他们用RfamGen的损失函数来近似对数概率ln P(x),因为VAE的损失函数是对数概率ln P(x)的下界。他们还对EVMutation进行了基准测试,EVMutation是一种基于Potts模型的变体效应预测器。通过这种比较,作者确认了RfamGen在预测变体效应方面的准确性与EVMutation相当。这些结果支持了RfamGen用于序列调整的潜力。
编译 | 曾全晨
审稿 | 王建民
参考资料
Sumi, S., Hamada, M. & Saito, H. Deep generative design of RNA family sequences. Nat Methods (2024).
https://doi.org/10.1038/s41592-023-02148-8
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢