
今天为大家介绍的是来自Rishita Anubhai团队的一篇论文。基础模型(FMs)通过学习大量未标记的数据,展现出在多种任务中的卓越性能。然而,为生物医学领域开发的基础模型大多是单模态的,即它们独立训练并用于单一任务,比如仅用于蛋白质序列、小分子结构或临床数据。为了克服这一局限性,作者提出了BioBRIDGE,这是一个参数效率高的学习框架,用于连接独立训练的单模态基础模型,建立多模态行为。BioBRIDGE通过利用知识图谱(KG)学习在一个单模态基础模型与另一个之间的转换,而无需微调任何底层的单模态基础模型。

先前的研究表明,基础模型(FMs)经过大量数据训练后,可以用于不同领域。在生物医学领域,FMs被训练用于处理科学文献中的文本语料库、蛋白质数据的序列和三维结构、分子图和SMILES字符串以及蛋白质交互数据的关系图形式。这些预先训练的生物医学FMs与之前在更小数据集上训练的方法相比,已取得显著进步。在训练中引入多模态数据可以进一步提高了FMs的性能,尤其是在少数/零次预测设置中。然而,在扩展到两种以上模态时,这一想法遇到了关键问题:(1)计算成本;(2)数据稀缺。BioBRIDGE与ImageBind不同,ImageBind将图像设为中心模态,并通过微调将所有其他编码器与图像对齐,作者所提出的BioBRIDGE保持所有单模态FMs固定,并学习连接这些单模态FMs。图1展示了该概念。具体来说,BioBRIDGE通过生物医学知识图谱(KGs)学习跨模态转换。这种方法是通过利用以下见解来建模的:(1)数据充足性。通常,收集单模态数据比收集来自两种模态的配对数据更容易。因此,与多模态编码器的联合训练相比,桥接在大规模单模态数据上训练的独立模型享有数据充足和效率的优势。(2)结构转换。多模态生物医学KG包含由头部和尾部生物医学实体及其关系组成的三元组所代表的结构信息。它涵盖了丰富的模态集合,如蛋白质、分子和疾病,使得全面的生物医学分析和机器学习成为可能。作者通过利用KG三元组中丰富的结构,对齐单模态FMs的嵌入空间,该步骤通过跨模态转换模型实现。
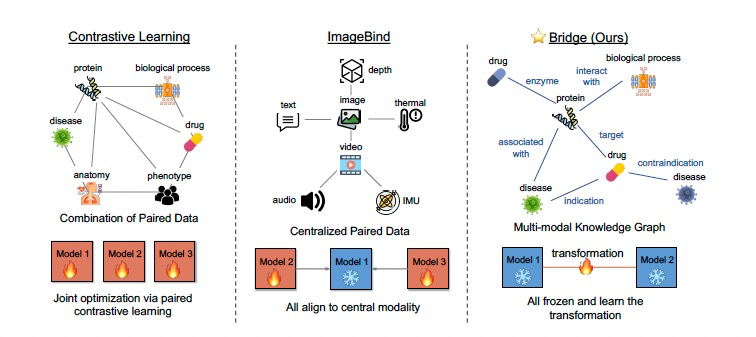
图 1
方法部分
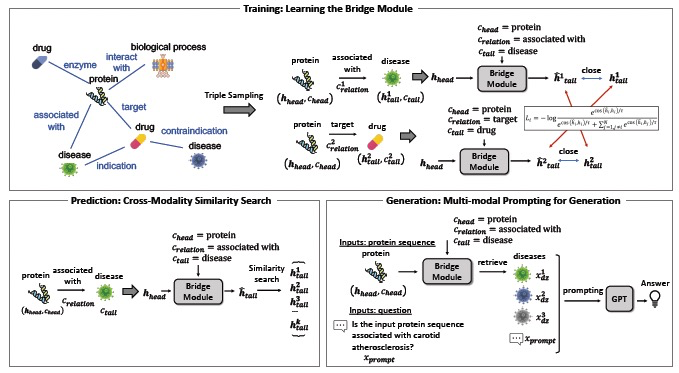
图 2. 模型流程部分如图2所示。
知识图谱(KG)由节点V和边E组成,表示为G = {V, E}。图中的一个节点,即vi = {xv, cv},其中cv是节点模态,xv是节点特征。例如,KG中的一个蛋白质节点,cv = “protein”,xv = 蛋白质的序列。连接两个节点vi和vj的边e ∈ E在知识图谱的上下文中被称为三元组,表示为tij = {vi, vj , rij},其中rij表示头节点和尾节点之间的关系。KG以分布式方式展示跨模态的关系,因此BioBRIDGE不需要设置中心模态。虽然传统的KG嵌入(KGE)方法也通过链接预测实现跨模态预测,但它们无法推广到训练KG中未包含的节点。相反,BioBRIDGE学习将头模态FM嵌入转换到尾部空间。在测试阶段不需要KG。有了在单模态数据上预训练的基础模型(FMs),作者的目标是桥接单模态FMs,以完成多模态任务,而无需微调FMs。对于来自不同模态的两个节点,作者将vi和vj编码为嵌入特征:hi = f(xvi) 和 hj = g(xvj),其中f(·) 和 g(·) 是两个FMs,因此hi和hj处于不同的空间。考虑到两个样本的模态类型及其关系,作者构建将hi投影到hj空间的转换对齐方法,并且可以通过基于嵌入特征的相似性搜索与来自跨模态的相关样本匹配。跨匹配方法通过对比学习InfoNCE损失函数实现。
作者从PrimeKG中选取了一部分子集来构建训练用的知识图谱。从图谱中挑选了六种主要的节点类型:蛋白质、分子、疾病、生物过程(BP)、分子功能(MF)和细胞组分(CC)。具体的训练集会根据下游评估数据集的不同而有所变化,以避免实验中的数据泄露。作者将六种类型的节点划分为三种模态:蛋白质序列、SMILES字符串和自然语言。技术上,作者使用ESM2-3B来编码蛋白质,使用UniMol来编码药物分子,以及使用PubMedBERT来编码疾病、生物过程、分子功能和细胞组分。对于文本节点,作者将其名称和定义连接起来,形成PubMedBERT的输入。
实验部分
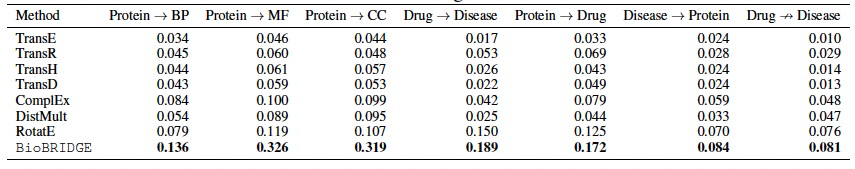
表 1
作者在表1中展示了七项跨模态检索任务的平均排名(MRR)。可以发现BioBRIDGE在知识图谱嵌入(KGE)方法中始终排名第一。专门的KGE算法从零开始学习节点和关系嵌入,仅基于KG,而作者的方法则建立在已经拥有丰富先验知识的预训练基础模型(FMs)之上。因此,BioBRIDGE以一种更加数据高效的方式连接模态。深入到不同任务的绩效中,如表1所示,作者观察到在KG中三元组较少的任务上,BioBRIDGE比基线有更高的优势。例如,“蛋白质→分子功能”任务中,BioBRIDGE比最好的基线高出约3倍,而在“蛋白质→生物过程”任务中,高出约1.6倍。这表明BioBRIDGE在有限数据下桥接FMs以完成多模态任务方面的优势,优于从零开始训练多模态模型。
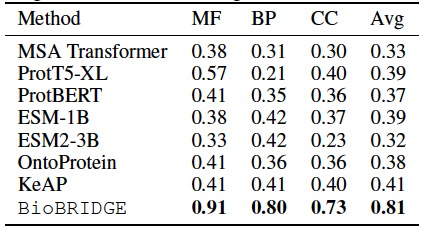
表 2
随后作者评估编码的蛋白质嵌入在捕捉生物分子功能相似性方面的能力,即生物过程(BP)、分子功能(MF)和细胞组分(CC)。作者遵循先前的基准测试方式,将蛋白质的基因本体(GO)术语注释作为目标,并使用转换到BP、MF和CC空间的蛋白质嵌入作为评估的输入。最终得分通过计算预测通过斯皮尔曼等级相关系数得出,数值越大越好。表2中报告了结果,作者的方法平均比最好的基线有约2倍的显著提升。在基线中,可以观察到通过KG增强的方法,包括KeAP和OntoProtein,比其他方法产生更好的结果,这表明连接蛋白质和生物属性的KG增强了蛋白质表示学习。然而,BioBRIDGE学会将蛋白质嵌入转换到生物分子功能嵌入空间,从而使蛋白质序列更好地与功能术语的语义含义对齐。此外,训练中涉及到的其他模态,如KG中的药物,进一步丰富了转换模型的监督。
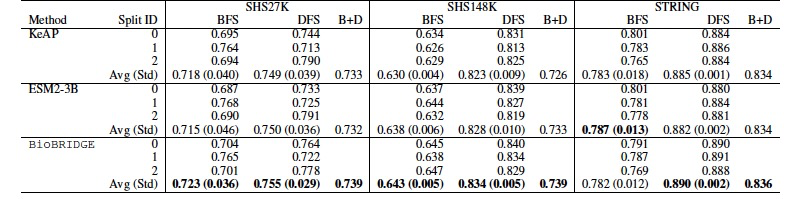
表 3
接着研究了蛋白质-蛋白质相互作用(PPI)预测任务。PPI预测任务的目标是对一对蛋白质的7种相互作用类型进行分类:反应、结合、翻译后修饰(ptmod)、激活、抑制、催化和表达。尽管PPI关系存在于PrimeKG中,但它仅代表物理相互作用(类似于七种类型中的“结合”),而其他六种类型属于领域外关系遵循先前基准测试的设置,作者使用预训练蛋白质模型提取蛋白质嵌入,作为要在PPI网络上训练的图神经网络模型的输入。作者的方法使用转换到蛋白质空间并带有ppi关系的蛋白质嵌入。作者报告了这个多类分类任务的F1分数。从表3中的结果来看,尽管结果在不同分割下有所变化,但作者的方法在大多数情况下都比基线有稳定的提升。结果表明,ESM2-3B的表现优于之前最先进的KeAP,这可以归因于其在庞大的蛋白质数据库上的预训练。BioBRIDGE通过注入“ppi”关系进一步增强了ESM2的嵌入。BioBRIDGE在样本较少的数据集(如SHS27K)上展现出更大的优势,因为它丰富了蛋白质嵌入与蛋白质-蛋白质相互作用本体信息。当训练数据量增加时,所有方法倾向于收敛到相同水平,而基线方法仍然不如BioBRIDGE。
编译 | 曾全晨
审稿 | 王建民
参考资料
Wang, Z., Wang, Z., Srinivasan, B., Ioannidis, V. N., Rangwala, H., & Anubhai, R. (2023). BioBridge: Bridging Biomedical Foundation Models via Knowledge Graph. arXiv preprint arXiv:2310.03320.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢