
今天为大家介绍的是来自Maria Duca团队的一篇论文。在化学生物学和药物发现领域,开发创新方法以识别RNA结合物引起了巨大关注。尽管针对细菌核糖体RNA的抗生素已经上市数十年,但对RNA靶向的重新关注反映了人们对更好地理解涉及RNA的复杂细胞内过程的需求。在这个背景下,小分子是用来探索RNA的生物学功能、验证RNA作为治疗靶点的工具,它们最终有可能成为新药。尽管近期取得了进展,但理性设计特定的RNA结合物需要更好地理解与RNA靶标发生的相互作用,以达到期望的生物学响应。在这篇综述中,作者讨论了接近这一未充分探索的化学空间的挑战。

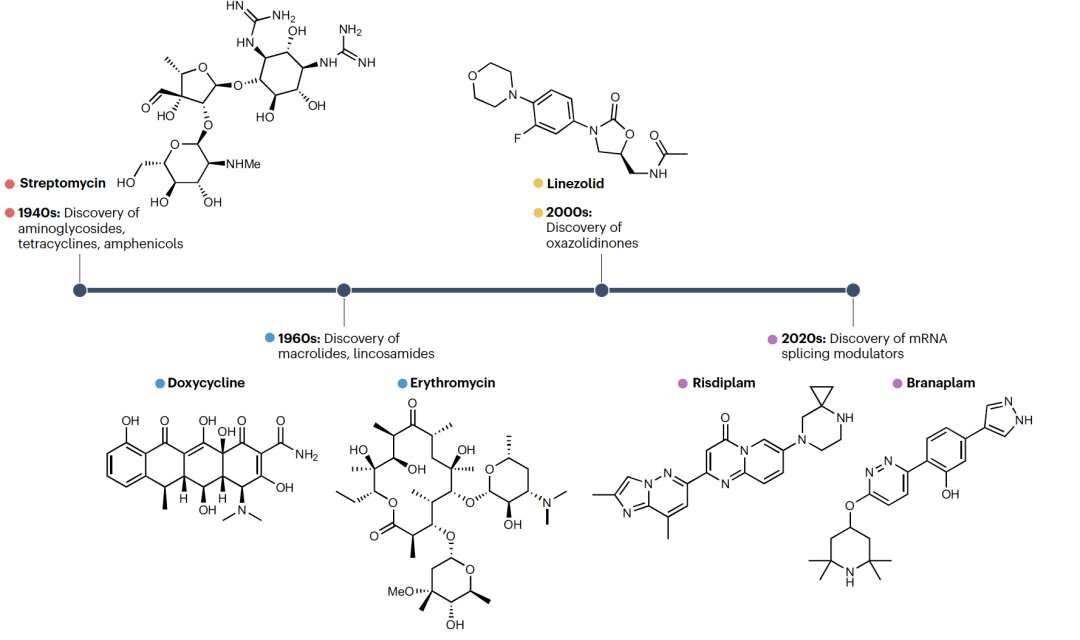
图 1
RNA靶向的范式,包括使用寡核苷酸和小分子,已经在化学和生物科学领域存在多年。认识到RNA在生命系统中的重要角色,导致了许多新RNA的发现,这些RNA不仅涉及转录和翻译,还涉及基因表达的调控。寡核苷酸通过序列互补性识别RNA。相比之下,小分子靶向结构化RNA中的特定位点,在这些位点中单链和双链区域的结合导致了特定三维结构的形成。这种结构识别依赖于与蛋白质中发现的结合口袋类似的结合口袋的形成。尽管寡核苷酸对RNA功能的基本研究和潜在治疗应用都是重要工具,但由于药代动力学和药效学的限制,例如稳定性、选择性和生物递送,它们的使用仍然受到限制。此外,由于RNA(特别是非编码RNA)高度结构化,寡核苷酸无法有效地接触或与这些结构相互作用。寡核苷酸的一些限制已得到解决,例如使用抗核酸酶的改良核苷酸来提高稳定性。然而,其他一些问题仍需解决,包括需要复杂给药途径的生物递送,因此应用性有限。与寡核苷酸互补的一种方法是使用小分子,这种方法对RNA功能的化学干预具有巨大潜力。第一种RNA结合剂是在20世纪40年代早期发现的,当时诸如氨基糖苷类抗生素链霉素等多种天然产物被识别为抗生素(图1)。抗菌活性是由于与原核生物核糖体RNA的结合,这导致了细菌中蛋白质合成的损害。随后,其他天然化合物被识别为具有抗菌特性的核糖体结合剂。这些包括四环素(如强力霉素)、大环内酯(如红霉素)和噁唑烷酮(如利奈唑胺)。RNA靶向的治疗潜力也最近被FDA批准的risdiplam(用于治疗脊髓性肌肉萎缩症)所凸显。
设计用于RNA结合的小分子
由于RNA的固有结构,识别RNA结合剂可能具有挑战性。在这方面的第一个障碍是高度负电荷的主链,这限制了兼容结构的数量,并且更倾向于与带正电荷的物种相互作用。此外,与蛋白质的20种氨基酸相比,RNA只由四种核碱基组成,这使得选择合适的化学支架成为设计适合RNA结合的配体的关键步骤。不同于大多数蛋白质靶标,生物学相关的RNA存在于一个动态的结构集合中,这取决于碱基配对模式。诸如盐浓度、细胞内相互作用或突变等因素影响某些亚结构的普遍性。改变这个动态集合可能会影响RNA的功能,确定哪种RNA构象在体内是有生物学活性的仍然具有挑战性。预测化合物将与之结合的结构异构体以及这将如何影响RNA功能的困难,是第三个主要缺点。幸运的是,结构生物学家现在除了X射线之外,还有一系列技术可用,如先进的核磁共振(NMR)和冷冻电镜,这些技术应该为未来的重要发展提供工具。鉴于这些前提,选择性RNA结合剂作为化学探针或潜在药物的设计不能以与蛋白质靶向相同的方式进行。事实上,小分子与RNA的相互作用主要由氢键和π相互作用主导,这构成了已报告的结合剂中小分子与RNA识别的一半以上的相互作用。疏水相互作用和弱氢键也是受青睐的相互作用,即使它们发生的频率低。相反,小分子与蛋白质复合物中涉及的相互作用主要由疏水接触表示,其次是氢键和π堆积。从发现第一个RNA结合小分子到最近开发出特异性和类药分子的领域进展,可能存在克服上述挑战的技术。然而,对特定RNA靶标进行配体的理性设计仍然困难。
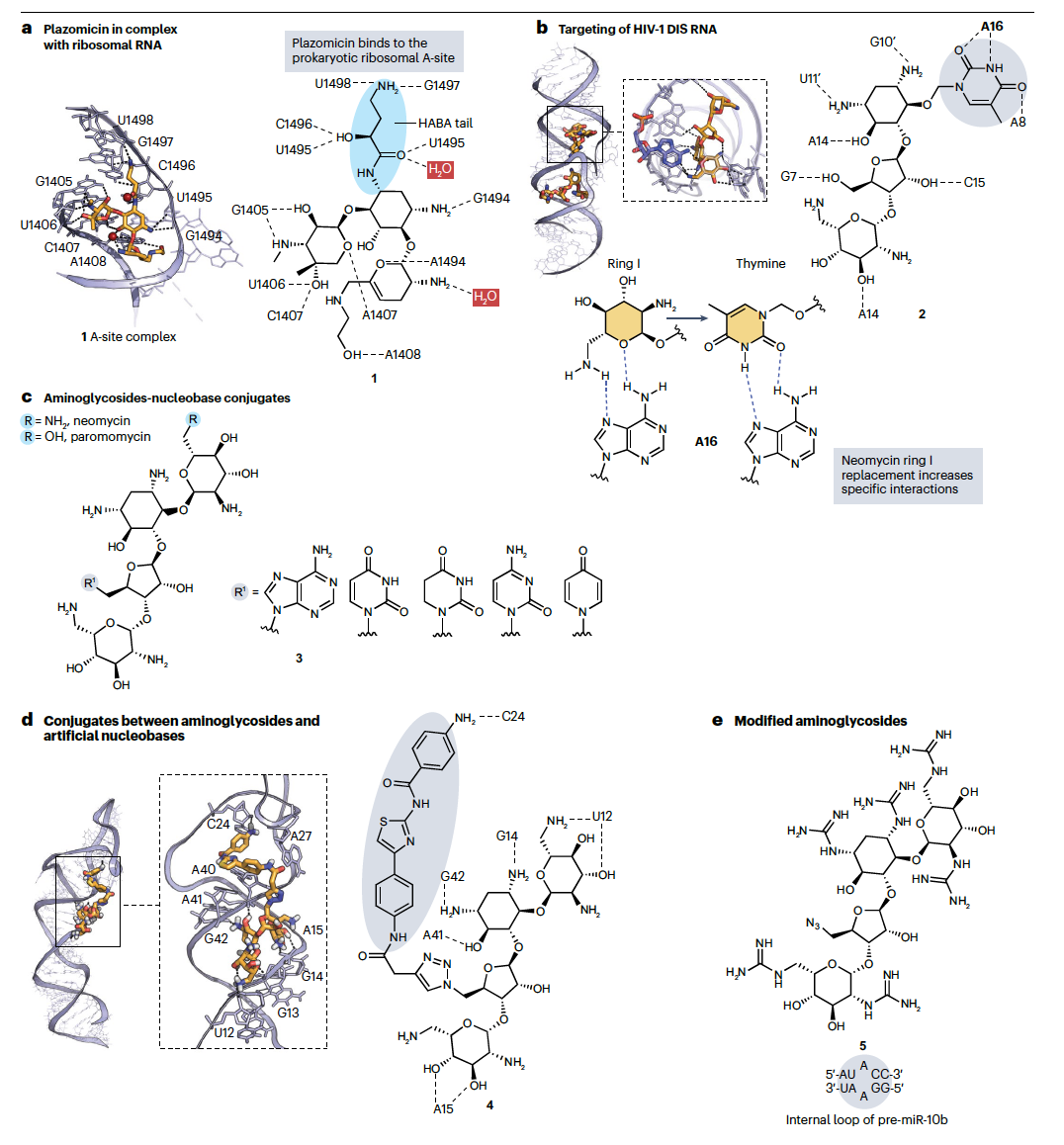
图 2
RNA的负电荷磷酸骨架支持使用在生理pH值下质子化的化学基团(如胺或胍)的化合物,这些化合物可以与磷酸提供静电相互作用和与未配对碱基形成氢键。然而,这些静电相互作用本身可能促进与多个靶标的非选择性结合。为了解决这个问题,可以考虑RNA上成对和未成对区域的存在,因为它们导致与双螺旋DNA不同、使得RNA更类似于蛋白质的三维结构。氨基糖苷类通过主要结合到内部环和隆起的RNA区域来阐明这一概念,这些区域扭曲了双螺旋结构。例如,plazomicin(化合物1,图2a右侧),一种最近获批用于治疗多重耐药菌引起的感染的氨基糖苷类,通过与核糖体A位点的π和氢键相互作用来与核碱基相互作用,这一点通过X射线揭示(图2a左侧)。核糖体A位点是与进入的tRNA结合的区域,将新的氨基酸引入正在生长的肽链中。此外,plazomicin的羟基-氨基丁酸尾部与尿嘧啶碱基形成了氢键。这导致即使形成了静电相互作用,也能产生强烈和选择性的相互作用,这些静电相互作用加强了结合,但妨碍了氨基糖苷类的特异性。为了进一步增加选择性,氨基糖苷类已与能够与靶标建立特定相互作用的基团结合。这是通过与核碱基结合来完成的,例如改良的新霉素,就是为了选择性地靶向人类免疫缺陷病毒-1(HIV-1)二聚化启动位点(DIS)。这是位于病毒基因组5'非编码区的一个高度保守的RNA茎环结构。其二聚化是HIV复制的关键步骤,其抑制已被考虑用于潜在的抗病毒治疗。新霉素与DIS复合体的结构研究强调了一个腺嘌呤碱基参与了与新霉素环I的关键相互作用。与A位点的比较显示了在这种情况下这种相互作用是不存在的。因此,环I被嘧啶替代,导致新霉素衍生物2(图2b)能够与DIS结合,与新霉素相比具有更好的特异性。这个例子说明了如何通过适当添加与RNA靶标结构互补的氢键供体和受体来增加选择性。类似地,新霉素和paromomycin与核碱基结合成为化合物3(图2c),以获得对HIV-1转录激活反应(TAR)RNA的亲和力比对A位点更好的配体。这个结果很难预测,因为即使在有结构信息的情况下,理性药物设计仍然经常具有挑战性,这可能是由于RNA系统本质上的动态性。
其他类型的杂环化合物已被用于准备针对致癌miRNA生物发生的氨基糖苷类结合物,这些miRNA是基因表达的重要调节剂。miRNA的生物发生经历两个高度结构化的前体:约1千碱基的原初miRNA(pri-miRNA)和miRNA的前体(pre-miRNA),它们被核糖核酸酶Drosha在细胞核和Dicer在细胞质中依次切割,产生约22-24个核苷酸的成熟miRNA。后者可以与mRNA结合并抑制其翻译成蛋白质。一些miRNA的过表达似乎是导致各种癌症的发展和进展的原因,这些致癌miRNA不仅成为不同种类癌症的生物标志物,也成为潜在的抗癌靶标。为了设计选择性结合致癌miRNA茎环结构前体并抑制miRNA生物发生的配体,新霉素与人工核碱基结合,能够与DNA和RNA碱基对形成特定氢键,形成碱基三元素。这导致了诸如化合物4(图2d)的化合物,它选择性地抑制癌细胞中过表达的一小组致癌miRNA的生物发生。值得注意的是,苯并噻唑杂环组分的结合使新霉素定向到致癌miR-372的茎环前体pre-miR-372,抑制Dicer切割和miRNA成熟。这导致有效地体外抑制miR-372并在过表达miR-372的癌细胞中特异性抑制增殖,没有毒性。酶促足迹分析(分析RNA序列中哪些残基受到配体存在时核酸酶切割的保护)和分子对接分析表明,加入第三个RNA结合域可以提高亲和力和选择性。因此,作为天然RNA结合剂的氨基酸被结合到新霉素-核碱基衍生物中,提高了结合和抑制活性。值得注意的是,尽管这些配体选择性地与pre-miRNAs而非tRNA和DNA结合,但它们也与其他一小组pre-miRNAs结合。表明选择性在很大程度上取决于细胞内靶标表达水平以及结合RNA的功能位点,以有效地抑制其功能。强烈结合到非功能性非靶点位点可能不会导致任何生物活性或毒性。因此,将结合与功能联系起来是设计具有生物活性、选择性RNA结合剂和评估潜在非靶标影响的关键。
离子化合物以选择性方式与RNA相互作用,提供对结合重要的静电相互作用。这些类型化合物的结合,能够形成特定氢键和疏水相互作用的基团,增加了选择性。因此,为了提高选择性,可以使用阳离子化合物靠近RNA磷酸骨架,同时与能够建立特定氢键或疏水相互作用的基团结合。
针对二级和三级RNA结构的靶向
发现RNA结合剂的第二个挑战是RNA靶标的特殊化学空间,这与蛋白质的不同。这需要一个新的范式来设计选择性配体,使用的策略可以大致分为两类:(1)基于识别可能与RNA结构形成的特定相互作用的理性设计配体;(2)使用高通量方法结合筛选、生物信息学和生物物理学来识别新的支架。这些策略的成功在很大程度上依赖于对目标的二级和三级结构的深入了解,这在确定配体的潜在结合口袋中起着关键作用。
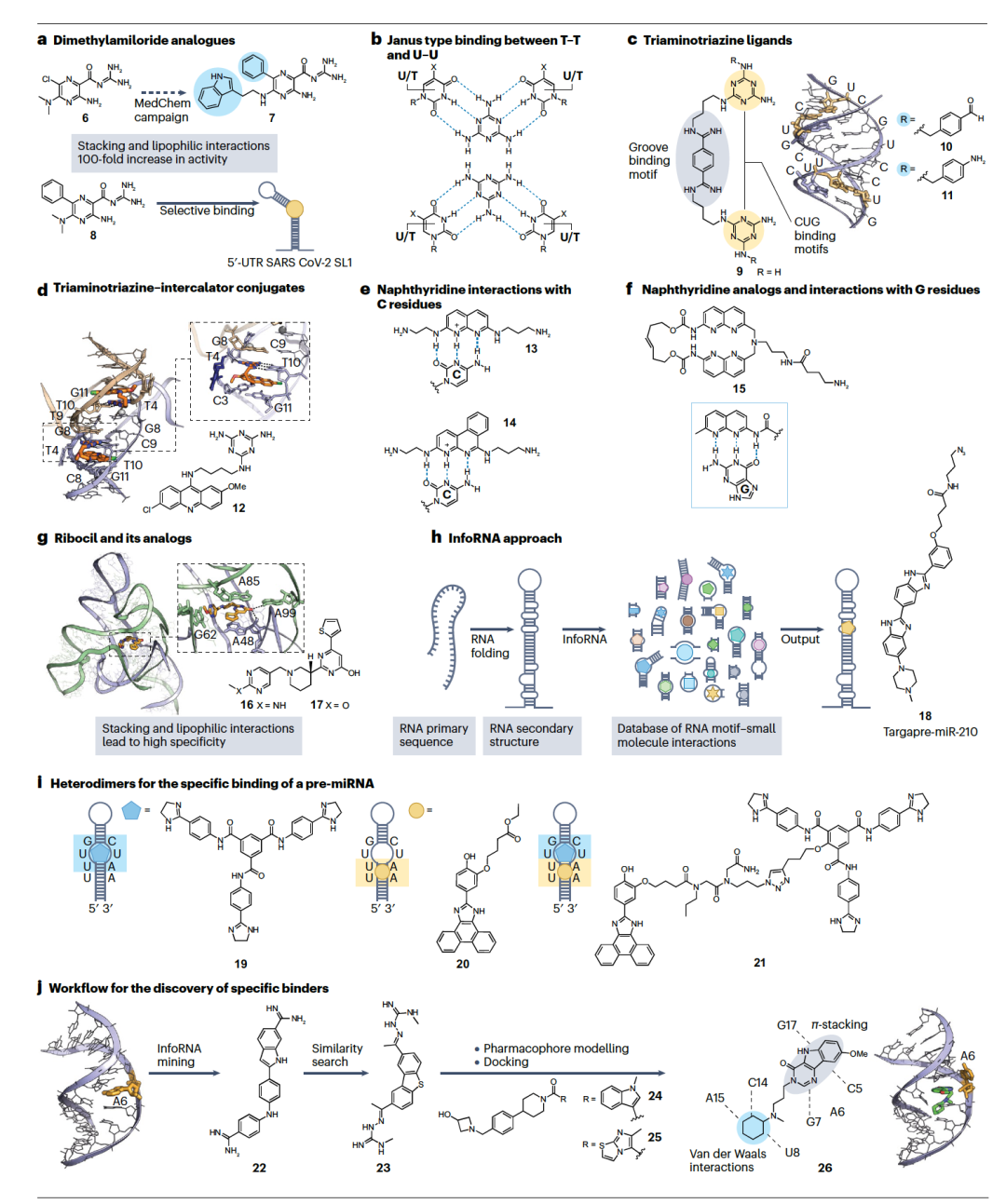
图 3
对于第一种策略,一个相关的例子是二甲基阿米洛利德(图3a中的化合物6),与氨基糖苷类相比,它显示出较低的亲和力但有前途的选择性。在C5和C6位置的反复修饰使其能够与HIV-1 TAR RNA的隆起区域建立强相互作用。衍生物7(图3a)在Tat肽置换测定中比原始化合物6的活性提高了100倍,这表明在Tat相互作用的特别重要位置有更强的结合。同一支架被修改用于针对其他病毒RNA。一些二甲基阿米洛利德类似物,如化合物8(图3a),通过减少SARS-CoV-2 RNA基因组的5′-未翻译区域域的隆起和/或环残基与特定结合位点的相互作用,展现出有前途的抗病毒活性。阿米洛利德类化合物因此可能是理解SARS-CoV-2病理的有希望的化学探针,即使它们通常表现出适度的选择性,因为它们也能够结合其他RNA或病毒蛋白。类似的相互作用也发现于三氨基三嗪部分,它已被用于设计各种DNA和RNA配体,并且对T-T和U-U错配的识别负责。嗪杂环的两个边缘可以与配对不良的尿嘧啶和胸腺嘧啶残基形成一整套Janus楔形氢键(图3b),或者可以与两个核碱基中的一个结合,并将另一个从螺旋中排出。在化合物9-11(图3c)中添加了双酰胺基连接器,使得准备用于靶向DNA和RNA三联体重复扩张的多聚化合物成为可能,这些由DNA中三个或更多核苷酸的异常重复以及随之而来的RNA中表示。这些与一些遗传疾病有关,因为它们诱导异常蛋白的产生。酰胺基团的存在还增加了与双螺旋主沟中磷酸骨架的静电相互作用。
基于高通量技术,一项显著的研究报告称将高通量测序与传统的SELEX方法结合,随后进行生物信息学分析,用于识别pre-miRNA的发夹环序列,显示出对水溶性循环错配结合配体的高结合亲和力和特异性,该配体能够选择性地与未配对的G残基相互作用(图3f)。观察到内源性pre-miR33a和包含特征性结合基序的pre-miR24-2的结合。在pre-miR33a的体外Dicer切割反应中,也观察到在化合物15存在下剪切产物的剂量依赖性减少,从而证实了这种方法的潜力,尽管未报告细胞内活性。尽管化学上与氨基糖苷类不同,鸟苷化和质子化化合物仍然保持基于静电力的相互作用,这可能会损害选择性。在这种情况下,ribocil(化合物16,图3g)标志着朝着具有药物样属性的特异性RNA结合剂的重大转变。作为黄素单核苷酸(FMN)核糖开关的抑制剂,ribocil靶向这些调节基因表达的保守原核生物RNA,这些RNA通过结合特定的内源性配体来调节基因表达。FMN核糖开关调节核黄素浓度,并控制与其生物合成和运输相关的基因。研究人员已经探索了各种类似物,包括ribocil D(化合物17,图3g),揭示了π堆积相互作用以及腺嘌呤48的羰基、2' -OH和腺嘌呤99的外环NH2之间的关键氢键。这表明,如果建立了氢键,RNA结合不严格要求质子化基团。
最新的结合方式
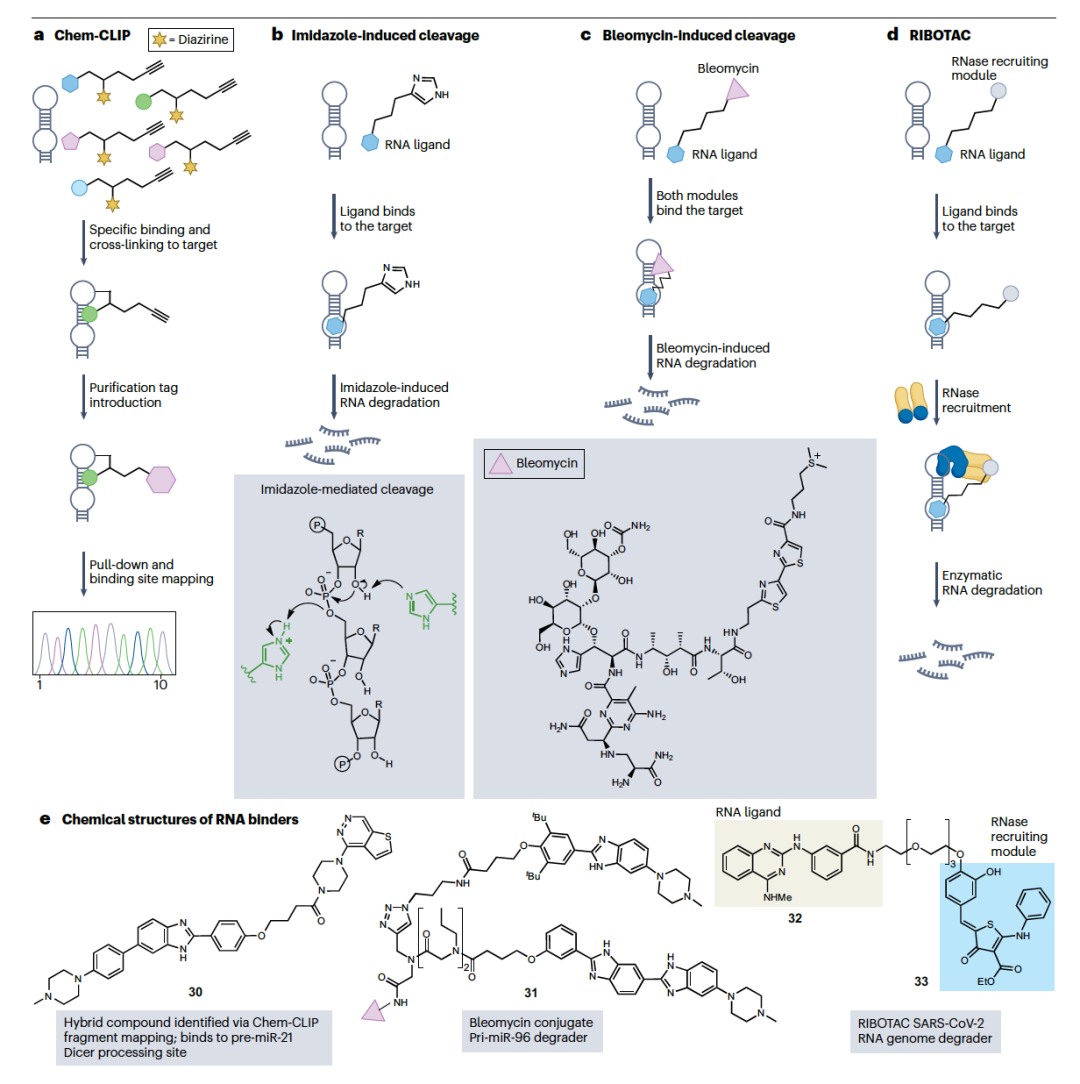
图 4
最近的策略使得设计具有增强特性的配体成为可能。在这方面一个有效的方法是Chem-CLIP。在这项技术中,一个RNA结合分子被配备有一个化学交联剂,以及一个生物素纯化标签(图5a)。RNA结合时,交联剂被带入高浓度并进行基于邻近的反应。通过生物素纯化的共价加合物揭示了分子的细胞内靶标。Chem-CLIP被用于绘制针对致癌前体miR-21的结合位点,从而识别出460个重氮化合物的片段,这些片段与RNA结合。值得注意的是,将有希望的片段与前体miR-21的结合剂结合,得到了化合物30(图5b),该化合物在乳腺癌细胞中抑制了前体miR-21的加工。共价RNA配体结合为配体发现和细胞内RNA靶标验证提供了一种有前途的方法。联合转录组宽分析可以进一步阐明细胞内靶标,正如PreQ1核糖开关和QSOX1 mRNA 5′-未翻译区结合剂的研究所证明的那样。
将切割活性与RNA结合结合起来是另一种策略,它通过永久损害RNA靶标来增加生物学活性。这种灵感来自于RNA酶,它们由于在催化位点中有一个或两个组氨酸残基而切割RNA。这些残基作为酸性和碱性的咪唑和咪唑基团,在过渡状态下与磷中间体协调,以及其他质子化侧链如赖氨酸(图4c),诱导磷酸二酯键的切割。在RNA配体中添加一个或多个组氨酸或咪唑残基,如插入剂、肽或氨基糖苷类,也被用作诱导RNA切割的策略,其特异性取决于RNA结合剂的选择性。bleomycin是一个已知能诱导DNA和RNA切割的化合物。它是从链霉菌中提取的天然产物,并被用作抗癌剂,具有复杂的作用机制,包括拓扑异构酶抑制。bleomycin也被应用于RNA切割,通过研究其优先切割位点并在miR-10b的前体中识别它。切割发生在Drosha和Dicer切割位点附近,这对成熟miRNA的产生至关重要,它在体外和细胞中被证明能够减少致癌miR-10b的产生。然后,bleomycin与特定RNA靶标的结合剂结合,如targapri-miR-96(化合物31,图4b)或r(CUG)exp的配体,以诱导特定的切割和靶标降解。总之,这些基于将共价或切割部分结合到RNA配体的例子说明了这些方法论不仅在靶标验证研究中的价值,而且在不影响选择性的情况下增加实际的生物学效果。
编译 | 曾全晨
审稿 | 王建民
参考资料
Kovachka, S., Panosetti, M., Grimaldi, B. et al. Small molecule approaches to targeting RNA. Nat Rev Chem (2024).
https://doi.org/10.1038/s41570-023-00569-9
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢