
今天为大家介绍的是来自Ali Madani团队的一篇论文。蛋白质语言模型从进化的多样序列中学习,已被证明是序列设计、变异效应预测和结构预测的强大工具。蛋白质语言模型的基础是什么,它们如何在蛋白质工程中应用呢?

蛋白质由一条线性的残基链组成,20种标准氨基酸构成了大多数天然蛋白质的词汇。这些氨基酸的排列顺序决定了蛋白质在其环境中的三级结构,进而赋予了它们特定的功能。理解蛋白质序列、结构和功能之间的关系是生物学研究的一个主要焦点。在这篇入门文章中,作者专注于一类仅基于序列操作却能捕捉到蛋白质结构和功能属性的机器学习模型。蛋白质语言模型(PLMs)是在覆盖生命进化树的大量蛋白质序列数据集上训练的。通过这些序列,PLMs学习到了蛋白质结构和功能的基础知识,使得它们能够完成广泛的蛋白质建模和设计任务。
蛋白质的进化
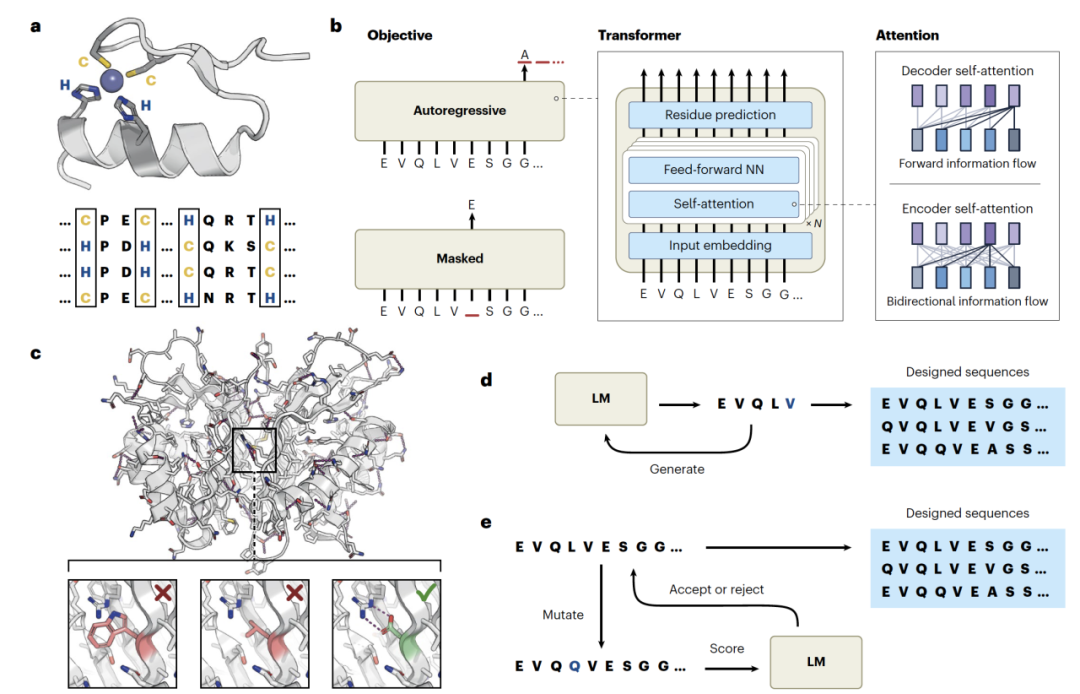
图 1
随着高通量DNA测序技术的进步,我们能够从各种各样的来源收集到数十亿个蛋白质序列。观测到的蛋白质序列数量(数十亿)的增长速度已经超过了结构数据收集(数十万)的速率。随着我们观测到越来越多的蛋白质序列,我们可以开始识别出驱动进化过程的模式。从结构角度看,某些突变可能通过破坏α-螺旋、引入未满足的氢键或埋藏带电原子,对二级或三级结构造成较大的干扰。一般来说,这些突变并不完全受限,但一般对现有折叠的维持倾向足够强。从功能角度看,特定的氨基酸(或残基)必须布局协调,以执行蛋白质的生物学角色。例如,在锌指基序的情况下,几个残基必须存在并且正确定位,以绑定金属离子(图1a)。就像结构约束一样,这些功能布局的违反可能发生,并产生新的功能性。然而,这样的事件极为罕见,意味着我们通常会观察到关键功能位置很少变化,如果发生这样的变化,它们通常会被其他位置的变化所补偿,这些变化共同定义了功能布局。这些对蛋白质序列的软约束通常被称为共进化。研究人员利用这些共进化信息已经促进了蛋白质建模的进步,尤其是对于蛋白质结构预测。通过语言模型,我们旨在明确地模拟蛋白质中残基之间的相互依赖性。
蛋白质语言模型的基础
根本上,蛋白质语言模型旨在预测给定迄今为止收集的所有蛋白质序列数据,观察到特定蛋白质序列S的可能性有多大。用S = (s1, s2,…, sN)表示一个蛋白质序列,其中si代表序列中位置i的氨基酸。作为初步估计,我们可能会考虑观察到一个蛋白质作为观察到其每个组成氨基酸的联合概率。在这个被称为unigram的模型下计算序列S的概率。实际上,为了计算P(S),我们只需统计我们的序列数据库中每个氨基酸发生的频率,并乘以特定序列S的概率。然而,蛋白质不是无序的氨基酸集合。相反,我们观察到的氨基酸的特定顺序是结构和功能的关键决定因素。为了捕捉这种顺序依赖性,我们可以使用前面的残基来预测下一个氨基酸的概率:
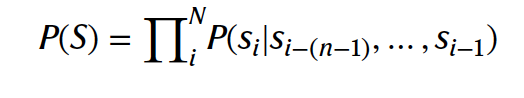
在一个n-gram模型中将这些上下文的概率相乘,形成序列的总概率。当n = 2时,这个模型被称为bigram,我们可以统计序列数据集中每个氨基酸在前一个氨基酸之后发生的频率来计算P(S)。Bigram模型可能能捕捉到二级结构的模式,这些结构显示出不同的氨基酸倾向性,但它不足以模拟由长段序列分隔的依赖性,正如图1a中的锌指域所示。为了捕捉长距离依赖性,我们可以简单地增加模型考虑的前置残基的数量。然而,实际上,随着模型扩展以考虑更多的上下文,需要通知统计测量的序列数量呈指数级增长。对于图1a中的锌指域,活性位点跨越21个连续残基,意味着我们观察到的特定排列是20的21次方种可能性之一。为了应对这一挑战,现代语言模型通常使用一种称为Transformer的神经网络架构,它能够从数据学习任意长度上下文的序列依赖性。
Transformers将整个序列上下文纳入考虑
Transformer模型最初是为了自然语言的机器翻译——例如,将英文文本翻译成德文——而提出的。原始的Transformer模型包括一个编码器,用于总结源文本,以及一个解码器,用于生成目标语言的文本。然而,在自然语言处理和蛋白质序列建模的许多应用中,这些组件被单独作为仅编码器和仅解码器语言模型使用。这两种类型的模型的网络架构大体相同(见图1b)。首先,氨基酸的输入序列被一个输入嵌入层投影到一个“隐藏的”或潜在的序列上。接下来,一系列重复的注意力层(见下文)和前馈网络处理序列表示。最后,一个残基预测层将处理过的序列表示投影回到氨基酸的预测分布上。最终,模型被训练来填补序列末尾(仅解码器)或序列中间(仅编码器)的缺失氨基酸。Transformer的关键创新之一是使用注意力机制来模拟序列间的全局依赖性。直观上,注意力机制使模型能够学习序列上下文的哪些部分对给定的预测是相关的,就像人在被问到一个阅读理解问题时可能会比其他部分更关注文章的特定部分一样。当一个序列表示传递到一个注意力层时,每个位置会发出一组查询和键向量。如果来自某一位置i的查询与另一位置j的键匹配(通常通过点积来衡量),网络就会从i到j分配高度的注意力。所有位置对的注意力值被收集到一个维度为N×N的注意力矩阵中。每个位置也会发出一个值向量。为了更新序列表示,我们根据从i到所有其他位置j的注意力及其各自的值向量,计算每个位置i的加权和。实践中,训练在蛋白质序列上的仅编码器和仅解码器模型已被证明相当有用。仅编码器模型通常用于学习序列的表示,然后适应于各种下游任务,而仅解码器模型用于生成和评分蛋白质序列。
自回归语言模型生成和评分蛋白质
解码器模型有时被称为自回归语言模型,因为它们通过迭代地基于之前的输出预测下一个残基的方式进行训练,从而生成序列。它们通过下一个令牌(token)预测目标进行训练,其中下一个氨基酸的概率是由整个前序序列信息决定的:我们在一个序列数据库上训练自回归模型来预测P(si|s<i)。为了促进这项任务,使用因果掩码来限制模型中的注意力操作,使信息只能从较早的位置流向较晚的位置,而不能反向流动(见图1b,解码器自注意力)。用于蛋白质序列的著名自回归模型包括unirep、progen和protgpt2。自回归模型可以生成采用各种折叠的多样化序列,而且预测的p(s)也已被证明与蛋白质的功能适应性相关。序列是通过从预测的分布p(si |="" s<i)中迭代抽样下一个残基生成的,每个抽样的残基被附加到序列上以通知后续的预测。以类似的方式,可以通过根据模型计算序列p(s)的可能性来评分序列,这可以被认为是给定序列被训练数据中的进化上产生的可能性。这些模型已在包括基因组、宏基因组和免疫库序列在内的各种数据集上进行了训练。通过修改训练数据的组成,我们可以改变模型生成的序列类型,以及学习更好的适应性预测器。训练数据与模型预期应用之间的这种一致性是一个关键考虑因素,对性能有重大影响。<="" p="">
掩蔽语言模型学习可泛化表示
对于编码器模型,训练目标被修改为预测序列中各残基的token。具体来说,随机选择一部分残基并用一个特殊的掩蔽token替换,然后模型(称为掩蔽语言模型)的任务是预测它们的token。与自回归模型不同,掩蔽语言模型使用双向注意力,并考虑序列中的所有残基来进行预测(见图1b,编码器自注意力)。蛋白质序列的著名掩蔽语言模型示例包括ESM和ProtTrans模型家族。为了在掩蔽语言建模目标上表现良好,模型必须学习一系列广泛的蛋白质特征。例如,为了预测一个被掩蔽残基的身份,模型被隐含地鼓励(即,无需监督)构建二级和三级结构表示(见图1c)。掩蔽语言模型的注意力矩阵也被证明可以直接编码蛋白质结构,以残基-残基接触图的形式。除了结构特征外,掩蔽蛋白质语言模型还捕捉到生物物理特性、进化上下文和家族内的排列。由于它们学习了可泛化的表示,掩蔽语言模型经常被用来为多种下游序列预测任务编码给定的蛋白质,如功能活性或相互作用的预测。
生成和优化功能性蛋白质
Madani等人(2023年)使用语言模型生成功能性蛋白酶。一个拥有超过10亿参数的自回归语言模型在超过280百万蛋白质序列上进行了训练,这些序列来自于超过19,000个家族。训练过程中加入了从给定蛋白质的相关元数据中衍生的标签,以实现高效学习,并主要提供一种可控制的——即条件性的——从期望输入参数生成序列的方法(例如,生成一个人工序列库,这些序列很可能属于预定义的蛋白质家族)。通过迭代抽样下一个氨基酸,并将之前抽样的残基上下文作为输入喂给模型(见图1d),生成了超过一百万的人工序列。为了改进序列的多样性和质量,已经开发了多种语言模型的解码策略,包括束搜索(beam search)、top-k抽样和nucleus抽样。这些技术中的每一种都在自回归解码的每一步重新塑造概率分布,平衡计算成本与生成序列的多样性和质量。与从头生成序列相比,大多数蛋白质工程努力旨在优化蛋白质的功能,前提是可以获得高度可靠的测定方法。在这种情况下,起点或父序列是已知的,并通过定向进化进行迭代优化。语言模型可以在监督设置中训练,使用来自实验数据的序列-标签对。Biswas等人(2021年)使用至少24个功能性测定的突变序列训练了一个带有监督语言模型的适应性预测器。使用马尔可夫链蒙特卡罗程序优化绿色荧光蛋白和β-内酰胺酶的序列(见图1e)。在马尔可夫链蒙特卡罗建模中,生成随机突变,通过PLM对结果蛋白质的可能性进行评分,并根据可能性接受或拒绝提议的突变。这些在计算机中设计的序列已被证明在湿实验室中具有改进的功能性。蛋白质语言模型在生成功能性蛋白质和促进给定蛋白质的优化方面已被证明是有效的。展望未来,可控制地生成功能特定的蛋白质序列仍是一个充满希望的领域。当前技术需要在一组精选的自然蛋白质上进行微调,这对于代表性不强的家族或新颖功能来说可能是一个挑战。消除这一限制可能会实现功能性蛋白质的按需生成。
编译 | 曾全晨
审稿 | 王建民
参考资料
Ruffolo, J.A., Madani, A. Designing proteins with language models. Nat Biotechnol 42, 200–202 (2024).
https://doi.org/10.1038/s41587-024-02123-4
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢